StereoMiner
StereoMiner
时空组学高级分析在DCS智能云平台完成SAW标准分析后,可进行0代码高级分析,多种参数灵活配置,分析结果直观呈现,可一键投递全流程任务。主要流程包括数据预处理、聚类/注释、拟时序分析、互作分析、功能富集分析,并最终形成高级分析报告,完成时空分析交付。
单细胞组学高级分析的主要流程同时空组学类似,包括数据预处理、聚类/注释、拟时序分析、互作分析、功能富集分析,最终形成单细胞高级分析报告。
分析入口
1)交互分析入口
在交互分析页面,可根据需求选择“时空组学高级分析”或“单细胞组学高级分析”的入口进入。

2)任务入口(仅支持时空组学)
在DCS Cloud任务列表页,为每个完成时空SAW标准分析的T任务,提供高级分析入口。可点击“更多”中的“交互式工具”按钮,进入分析。

选择文件
- 时空组学高级分析
1) 点击“选择“按钮,进入数据管理窗口选择表达矩阵gef文件。可选择项目中已存在的gef文件,或自行上传gef文件至数据管理后再进行选择。

2)支持添加多个文件,选择binsize,同时投递任务。
提示
对于cellbin.gef,无需选择binsize,均只会投递cellbin的任务。

- 单细胞组学高级分析
1) 请选择上游单细胞流程输出结果所在的文件夹,该文件夹中需直接包含以下三个文件:“features.tsv.gz”、“barcodes.tsv.gz”、“matrix.mtx.gz”。

2) 注意:若使用云平台单细胞工作流“scRNA-seq-analysis-pipeline”输出的结果,请选择该任务结果路径下的“/.../04.Matrix/FilterMatrix/”文件夹(该矩阵为过滤后的有效数据),若选择其他文件夹可能导致结果错误。

3) 单细胞支持单个样本具有单文库或多文库运行分析,若一个样本中有多个文库,可点击矩阵文件夹后的“+”选取不同文库的数据。
提示
运行单细胞高级分析前,需手动填写每个样本的样本名称,用于区分后续单个样本的结果和任务信息。

- 若需同时运行多个样本的流程,可点击样本名称后“+”号添加多个样本的信息。

任务投递
可选择进行全流程投递或单步投递,每个投递方式均提供操作指引。


- 全流程投递:选择文件后,点击“全流程投递”,可进行流程选择、参数设置,提交后即可一次性跑完所选的全部流程(预处理和聚类或注释流程为必选)。

- 单步投递:选择文件后,点击“单步投递”,则只进行单步流程的投递,跑完质控任务后即停止,用户可查看结果并进行下一步投递。

任务结果查看
- 可对单细胞和时空高级分析完成后的某个任务进行查看,并在任务列表中切换流程结果。

- 任务列表呈树形结构,可体现流程之间的前后依赖关系。用户可由此判断该任务结果是基于哪个前置任务进行的,任务名可支持用户在任务管理中自定义区分。

- 若需查看其他芯片的分析结果,也可在load data界面更换文件,右侧任务列表将会同步该文件的历史任务,若无历史任务则列表为空。

质控流程
对gef文件的MID Count,Gene Type,线粒体gene比例等数据进行统计,该步骤预计30min(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(QC开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。

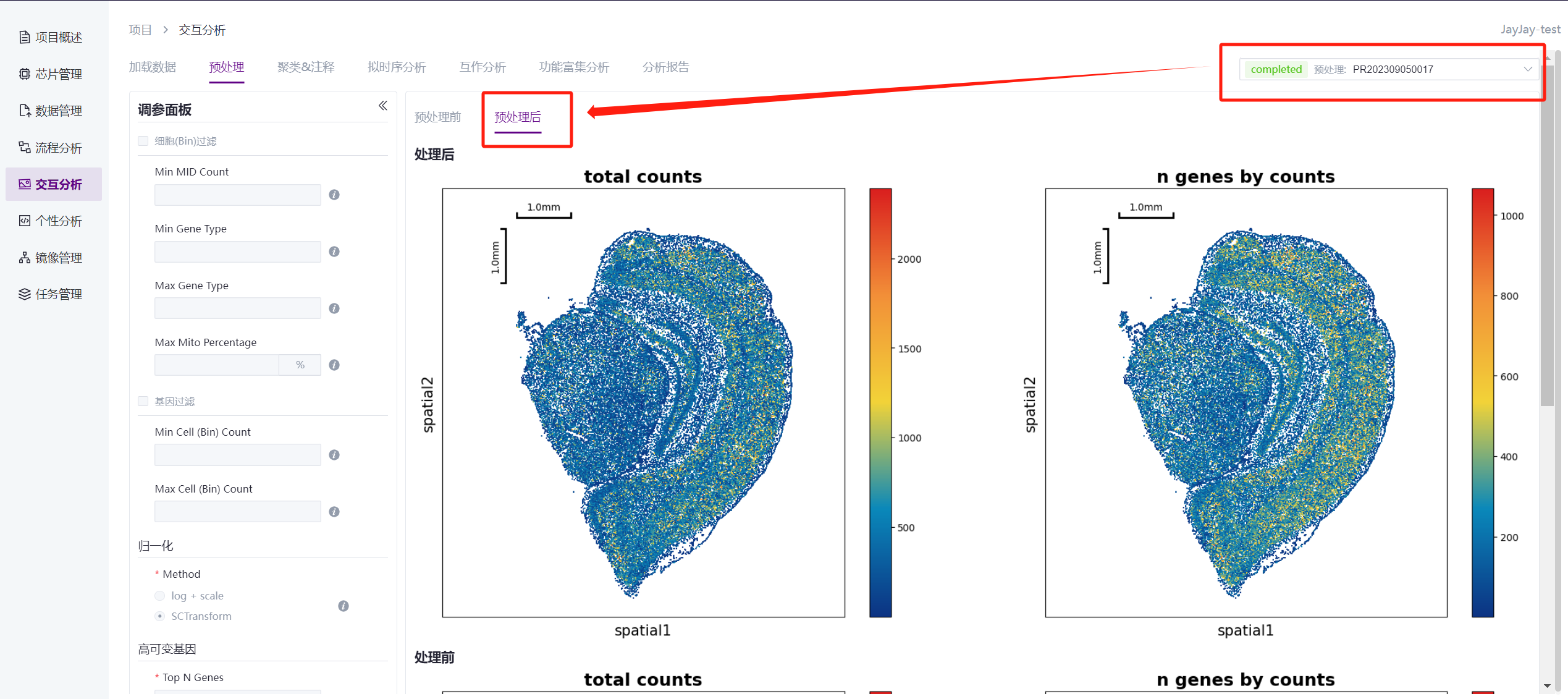
预处理流程
根据初步的统计结果,对gef文件进行细胞过滤,基因过滤,归一化,高可变基因等预处理分析。该步骤预计15min(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(PR开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。
注意
预处理分析算法调用的是stereopy工具算法。当同时使用Min MID Count 和 Top N Genes过滤时,容易把有效数据全部过滤,此时,系统会将两个参数定义为None。
注释方法选择
高级分析细胞注释支持分步注释和端到端注释。默认情况下,会为您选择分步注释。单细胞组学高级分析推荐使用分布注释,端到端注释无法生成UMAP图像。
- 分步注释:首先,使用聚类算法(目前支持使用Stereopy或Spateo软件的Leiden/Louvain)进行聚类,然后基于Marker基因或数据集自动或手动进行聚类标注。
- 端到端注释:直接使用软件注释(目前支持SingleR)。

分群
预处理完成后,进行分群(cluster)和找marker基因分析。该步骤预计35min(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(CL开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。
请注意:预处理完成之后,才可进行cluster和find marker分析
注:cluster和find marker分析算法调用的stereopy工具算法。
1)设置好cluster分析参数后,点击提交按钮进行分析。

2)任务提交之后,页面显示任务分析等待中。
3)分析完成后,可在任务结果页面查看该任务(CL开头的任务号),通过查看报告按钮,进入结果查看。可查看空间和UMAP可视化结果,单细胞高级分析只有UMAP可视化结果。


4)切换cluster,可查看不同cluster的全部marker gene。表格支持搜索、排序和下载。

- 通过数据集连接器,可查看在数据集中,marker gene对应的细胞类型,供手动注释进行参考。
注:Stereominer搜集了人和小鼠物种不同组织的200+数据集,并人工审核和处理,详细可访问:
https://www.stomics.tech/sap/stereominer/database/#/study/。

- 通过知识库映射器,可查看在知识库中,marker gene对应的细胞类型,供手动注释进行参考。
注:知识库是Stereominer数据库人工搜集的如panglaodb,cellmarker,Cell Taxonomy等数据库的内容,手动注释可参考。

7)查看marker基因表达热图。

自动注释
- 通过聚类分析得到不同细胞类群后,除了使用每个cluster的Marker基因信息对细胞类群进行初步的注释和分析之外,还可通过其他软件将细胞类群与参考数据库进行比较,以得到分群后不同细胞的细胞类群注释信息。

SingleR 可以将细胞聚类结果中的每个cluster与参考数据库进行比较,根据该cluster与参考数据库中已知细胞类群基因表达谱的相似性对cluster进行打分,最终得到一个打分矩阵。通过打分矩阵结果即可将细胞聚类结果注释为不同的细胞类群。 该步骤预计60min(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(AA开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。
参考数据集有三种途径可供选择:
- SingleR自带数据集:选择“Default”,下拉选择人/鼠对应的参考数据。

- 知识库数据集:选择“Stereo Database”,可根据所提供的信息选择知识库内包含的数据。

- 数据管理:选择“Data Management”,可进入数据管理页面选择自定义参考数据。(注:请先下载模板文件,自行构建参考数据后上传至数据管理。)

也可跳过自动注释,分群完后直接进行手动注释的步骤。
手动注释
手动注释可在分群注释或自动注释后。若自动注释后进行手动注释,系统会将自动注释的结果展示在手动注释的第一列,可通过点击编辑按钮,进行细胞类型的修改,完成注释。手动注释完,也可重新进行分群或自动注释。请注意,若重新进行自动注释,手动注释的结果会被重置。手动注释步骤预计20min(1*1cm芯片,1G reads)。
分析完成后,可在任务结果页面查看该任务(MA开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。
1)根据markergene和知识库dataset参考,手动对分群(cluster)进行细胞类型注释(支持编辑/删除操作,删除后将显示为“nan”)。编辑好不同cluster细胞类型后,点击提交按钮进行分析。

2)分析完成后,可在任务结果页面查看该任务(MA开头的任务号),通过查看报告按钮,进入结果查看。可查看空间和UMAP可视化结果。
3)查看不同分群的表达情况

拟时序分析
拟时序分析(pseudotime analysis)也称为细胞轨迹分析(trajectory inference),是指根据不同细胞类群之间其基因差异表达的情况,获得细胞谱系的发育结果,构建细胞随着一个虚拟时间顺序的变化轨迹,以此重现细胞随时间变化而变化的过程。
基于细胞注释的结果,使用Monocle3 软件进行拟时序分析,根据软件推测的发育轨迹进行细胞类群的可视化。支持用户选择部分分群进行任务投递。
该步骤预计1h(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(PT开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。

根据细胞发育轨迹结果中的节点信息,结合不同细胞类群中基因表达量的变化,Monocle3可以通过机器学习算法计算处于最早发育阶段的细胞,实现拟时序分析。

在不同的细胞类群聚类结果中,选取Monocle3计算得到的Top5/Top10/Top15的Marker基因,按照已获得的拟时序结果进行基因表达量的拟时序作图。

细胞互作分析
细胞注释完成后,可居于注释的数据进行细胞互作分析,支持CellChat和Cellphonedb分析。
CellChat 是一个能够从单细胞数据中定量推断和分析细胞间通信网络的工具,利用网络分析和模式识别方法预测细胞的主要信号输入和输出,以及这些细胞和信号如何协调功能。CellChatDB数据库整合了来自KEGG和最近研究中的信号分析信息,该数据库囊括了已知的配体-受体复合物组成,包括配体-受体多聚体复合物以及几类辅酶因子。可使用CellChat工具来鉴定时空数据中的配体/受体关系和细胞间的通讯分子,研究不同细胞类型之间的相互交流及通讯网络。
CellPhoneDB是一个受配体及其相互作用的数据库,有别于其他数据库的是CellPhoneDB考虑了受配体的亚单位结构,准确的表达了异聚体复合物。
该步骤预计3h(1*1cm芯片,1G reads)。分析完成后,可在任务结果页面查看该任务(IN开头的任务号),通过查看报告按钮,进入结果查看。也可下载分析结果数据。

功能富集
细胞注释完成后,可基于注释的数据进行功能富集分析,使用clusterProfiler 软件分别对每一个细胞类群的差异基因进行GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)功能富集分析。该步骤预计0.5h(1*1cm芯片,1G reads)

横轴为富集基因占比,纵轴为富集到的GOterm,颜色对应P-value的值,越红表示富集效果越显著。

横轴为富集基因占比,纵轴为富集到的KEGGPathway,颜色对应P-value的值,越红表示富集效果越显著。

圆点为富集到的GOterm,圆点大小表示基因数,圆点颜色表示P-value的值,越红表示富集效果越显著;连接线的粗细表示两个GOterms之间共有的基 因数。

圆点为富集到的KEGGPathway,圆点大小表示基因数,圆点颜色表示P-value的值,越红表示富集效果越显著;连接线的粗细表示两个GOterms之间共有的基因数
分析参数说明
| 调参模块 | 参数名称 | 中文说明 | Description in English | 参数默认值 | 参数范围 | |
|---|---|---|---|---|---|---|
| preprocessing | 细胞(Bin)过滤 Cell (Bin) Filter | Min MID Count | 单个细胞(或bin)中所含的最小MID数。 | Minimum value of MID count expressed in a single cell (or bin). | 20 | [0,gef中的max值] |
| Min Gene Type | 单个细胞(或bin)中所含的基因种类最小值。 | Minimum value of gene type expressed in single cell (or bin). | 1 | [0,gef中的max值] | ||
| Max Gene Type | 单个细胞(或bin)中所含的基因种类最大值。 | Maximum value of gene type expressed in a single cell (or bin). | None | [用户设置的Min Gene Type,gef中的max值] | ||
| Max Mito Percentage | 单个细胞(或bin)中线粒体基因所占比例的最大值。 | Maximum percentage of mitochondrial genes in a single cell (or bin). | None | [1,100] | ||
| 基因过滤/Gene Filter | Min Cell (Bin) Count | 单种基因至少在该数量的细胞(或bin)中有表达。 | Minimum number of cells (or bins) in which a type of gene expresses. | 3 | [1,100] | |
| Max Cell (Bin) Count | 单种基因至多在该数量的细胞(或bin)中有表达。 | Maximum number of cells (or bins) in which a type of gene expresses. | None | [用户设置的Min Bin Count,None] | ||
| 归一化/ Normalization | Method | 1. log+scale:在对数尺度上进行归一化。 2. SCTransform:利用正则化负二项分布进行归一化。 | 1. log+scale: First perform logarithmic transformation on the original matrix, and then use scale to normalize the matrix. 2. SCTransform: Use regularized negative binomial regression to normalize the matrix. | SCTransform | [log + scale, SCTransform] | |
| 高可变基因/Highly Variable Genes | Top N Genes | 保留的高变基因数量。样本所含基因数须不小于此值,否则会报错。 | The number of highly variable genes retained. The number of gene type contained in the sample must be no less than this value, otherwise it will report errors. | 3000 | [1000, 20000] | |
| Clustering | Clustering Method | Tool | - | - | Stereopy | [Stereopy, Spateo(SCC)] |
| Algorithm | - | - | leiden | [leiden, louvain] | ||
| Resolution | 控制聚类数量及精细程度的参数。值越大,聚类数量越多。 | The parameter that controls the number and specificity of clusters. A higher value will lead to more clusters. | 0.5 | [0.1,2] | ||
| Feature | 选择用于后续分析的基因子集。 | Choose a subset of genes used for the following analysis. | High variable genes | [All genes, High variable genes] | ||
| PCA | PCs Number | 使用PCA将数据降至较低维度后主成分的数量。 | The number of principal components to compute after using PCA to reduce the data to a lower dimensionality . | 50 | [2, 100] | |
| Dimensional Reduction | Method | - | - | UMAP | [UMAP, tSNE] | |
| PCs To Be Used | 从PCA中生成的用于降维的主成分数量。 | The number of principal components from PCA to be used for dimensional reduction. | 30 | [2, PCs Number] | ||
| Neighborhood Size | 构建每个数据点周围局部邻域的数据点数量。小的邻域尺寸可以创建更多局部特征和精细的数据表现,而较大的邻域尺寸可以创建更多全局特征和粗略的数据表现。 | The number of data points that are considered in the construction of the local neighborhood around each data point. A smaller value can create more local features and fine-grained details, while a larger value can create more global features and a more coarse-grained view. | 20 | [2, 100] | ||
| Auto annotation | Method | - | - | SingleR | [SingleR] | |
| Reference | 请先选择来源,再选择文件。 1.Default:包含算法中自带的reference。 2.StereoMiner Database:可选择知识库中的reference。 3.Data Management:可选择已自行上传至数据管理中的reference。支持h5ad和rds格式,可在下方下载模板。 | Please select the source first, then select a reference. 1. Default: Includes the references from SingleR's built-in database. 2. StereoMiner Database: Select references from StereoMiner Database. 3. Data Management: Select references already uploaded to data management. H5AD and RDS formats are supported, and templates can be downloaded below. | Source: Default Reference: HumanPrimaryCellAtlasData | [Default, Stereo Database, Data Management] | ||
| Feature | 选择一类基因进行注释。 de: 使用聚类间的差异表达基因。 sd: 使用聚类间的高可变基因。 all: 使用所有基因。 | Choose a type of gene to perform annotation. 1. de: Genes that are differentially expressed between labels 2. sd: Genes that are highly variable across labels 3. all: Not perform any feature selection. | de | [de,sd,all] | ||
| De.Method | 若选择“de”参数,计算差异表达基因的方法。 1. classic: 对数差异变化排序。 2. wilcox: Wilcoxon秩和检验。 3. t: Welch t检验。 | Select method (if choose de) for calculating differential expression gene. 1. classic: Log-fold changes ranked. 2. wilcox: Wilcoxon rank-sum test. 3. t: Welch t-test. | classic | [classic,wilcox,t] | ||
| Pseudotime analysis | Tool | - | - | monocle3 | [monocle3] | |
| Interaction analysis | Tool | - | - | CellChat | [CellChat,Cellphonedb] | |
| Reference | - | - | Human | [Human,Mouse] | ||
| Functional Enrichment | Tool | - | - | clusterProfiler | [clusterProfiler] | |
| Reference | - | - | Human | [Human,Mouse] | ||
| P-value | 用于选择显著差异表达的基因,并调整富集分析的p值截止值。 | The P-value is used to select significantly differentially expressed genes and adjust the P-value cutoff on enrichment analysis. | 0.05 | [0.001,0.01,0.05] | ||
| P AdjustMethod | - | - | BH | [holm", "hochberg", "hommel", "bonferroni", "BH", "BY", "fdr", "none" ] |
时空组学交付报告生成
- 仅时空组学高级分析支持生成报告,单细胞组学暂不支持。
- 语言可选中/英文。
- 支持自定义Logo。
- 可自选报告模块,对应报告目录下“分析结果” -> “4 数据分析”的各个子模块。默认全选。
注意
若gef文件非平台标准流程所产出,则无“质控数据结果统计”部分的数据。
- 支持选择生成报告所需的任务数据,系统会根据跳转时的当前任务,自动填充相关联的任务数据。若切换上级任务,下级任务的下拉列表也会自动过滤。

支持选择多张芯片同时生成报告。操作步骤如下:
- 1.从当前任务中进入报告模块,则当前芯片文件会被自动选中,并进行任务号填充。
- 2.点击+号选择新增文件。

- 3.支持通过文件类型、任务号、SN号、样本号、创建者筛选文件。任务号可在该芯片对应的高级分析任务列表中进行查看。(注意:仅运行过历史任务的文件才可被选择。)


- 4.注意:请确保多张芯片所包含的任务模块一致,否则将无法生成报告。
- 报告支持下载(.html文件)和打印(.pdf文件)。
注意
仅在打印为pdf时可生成页码及目录。

- 支持“文件整合”功能:所选的报告模块所生成的结果(除h5ad文件外),将全部整合到数据管理模块的下述路径中,方便用户快速整理与交付。
