Single-cell-multi-sample-anlysis标准分析流程说明书
第一章 产品信息
1.1 产品描述
单细胞转录组测序(Single-cell RNA Sequencing, scRNA-seq)是指在单细胞水平上进行高通量RNA测序,可以获得混杂样本测序(bulk RNA-seq)所无法得到的细胞异质性信息。scRNA-seq能够有效解决细胞异质性问题,通过检测单个细胞的表达谱,鉴定细胞类群,深入研究肿瘤转移侵袭、瘤内异质性、微环境重编程和耐药,近几年来已广泛应用于肿瘤,免疫,生长,发育等生物研究领域。近几年,scRNA-seq出现了爆发式的发展和普及,技术不断优化和创新,可以检测到更多的细胞,更高的分辨率,更全面的信息。 Single-cell-multi-sample-anlysis标准分析流程,即单细胞转录组多样本分析流程,使用Seurat软件识别与解释单细胞原始的表达矩阵中异质性的来源,该步骤主要包括细胞与基因的质量控制,降维与聚类,标记基因鉴定与差异分析等。使用SingleR软件对不同的细胞类群进行细胞类型注释,使用monocle3软件根据单细胞基因表达的相似性进行拟时序分析来说明细胞类群之间的关系,使用cellchat软件对不同类群细胞之间的互作关系进行探究,然后使用clusterProfiler软件对挑选出的基因集进行GO富集分析与KEGG通路分析,最后产出整体的分析结题报告。通过用户图形化界面进行样本输入及报告输出的全流程管理,使用方法简单,且支持并行化运行,缩短分析周期,达到快速、高效交付的目的。
1.2 注意事项
1)支持10X及C4技术下机数据质控后产出的包含(barcodes.tsv.gz, features.tsv.gz, and matrix.mtx.gz)矩阵的文件夹作为输入。 2)使用流程需自行准备包含样本-矩阵数据路径-分组信息的csv表格文件,按照指定列名和描述填写真实信息,否则会导致流程报错。 3)流程包含数据质控,多样本整合,聚类分析,细胞注释,拟时序分析、互作分析、富集分析并生成多样本分析报告全流程,可自定义选择是否运行对应步骤。
第二章 产品介绍
2.1 分析流程图
Single-cell-multi-sample-anlysis标准分析流程是基于DCS云平台系统开发的一款自动分析流程,其中包括质量控制、多样本整合、降维聚类、鉴定标志基因、细胞注释、拟时序分析、细胞通讯分析、功能富集以及报告生成:
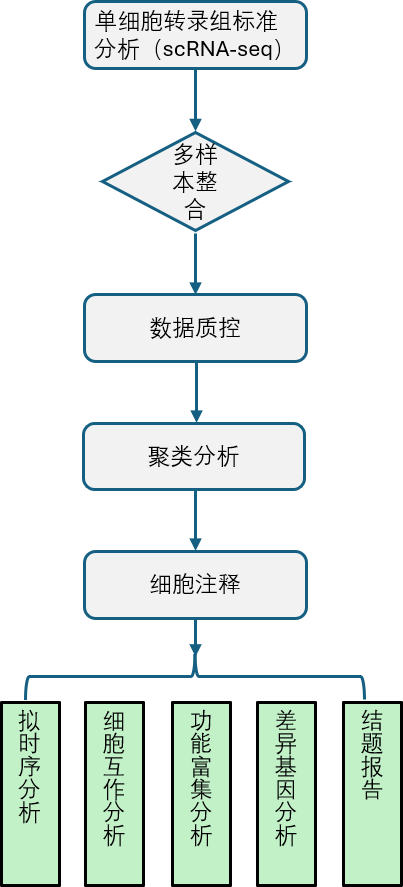
2.1.1 单样本质量控制
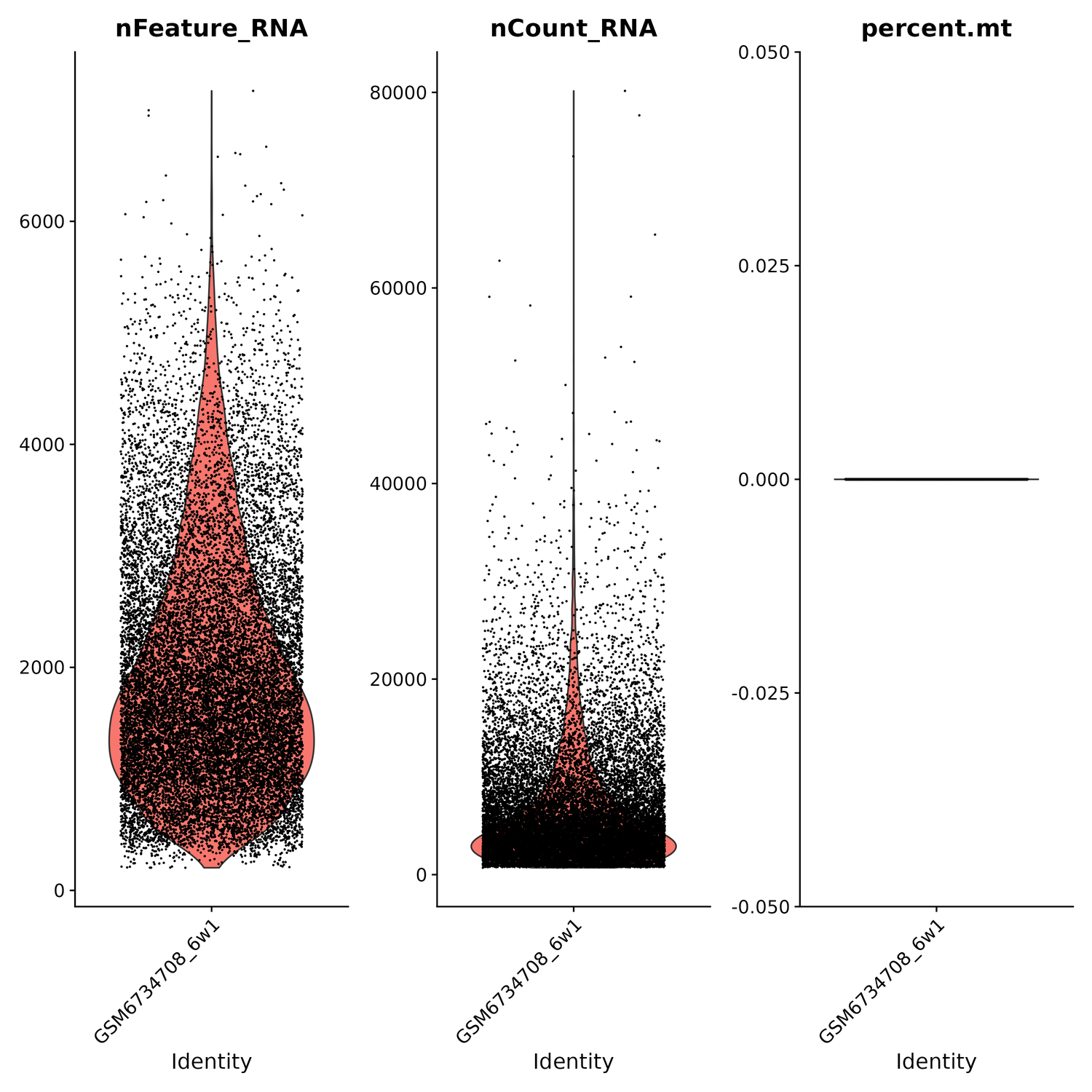
使用Seurat对每个矩阵文件进行严格的质量控制,包括数据过滤、标准化、鉴定高变基因和主成分分析,输出基因分布图、UMI分布和线粒体比例图、高变基因鉴定结果和主成分分析降维图。
2.1.2 多样本数据整合
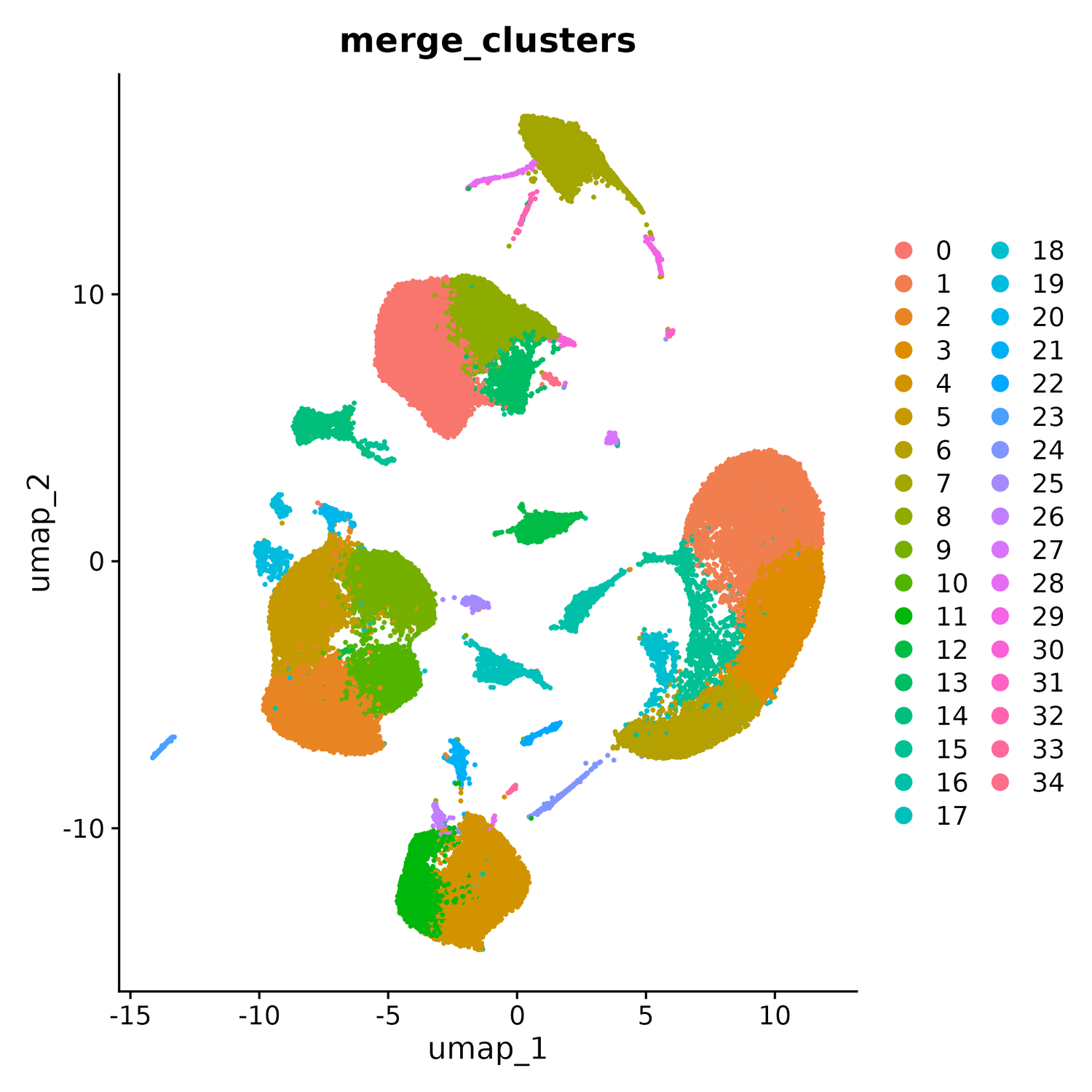
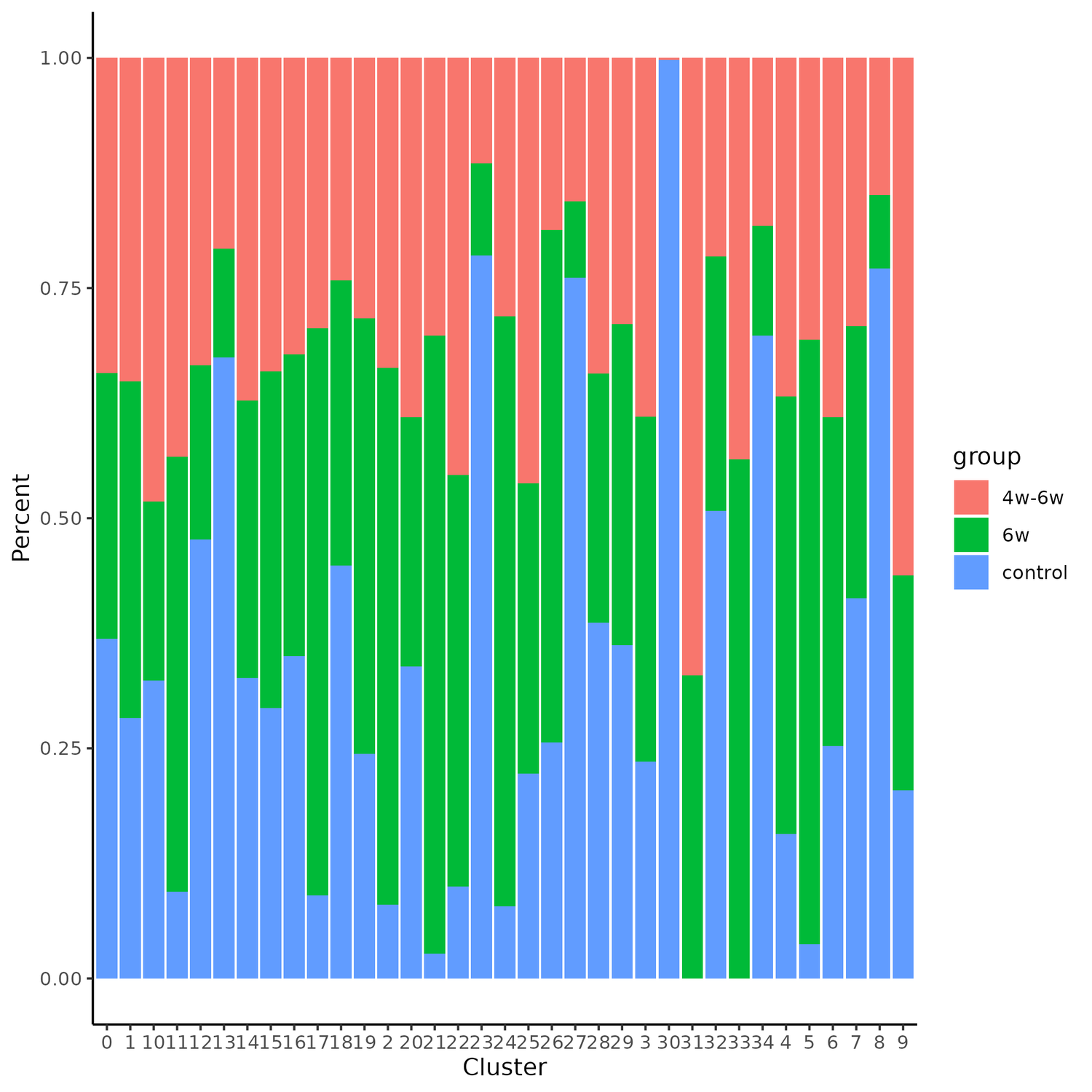
使用Seurat将所有矩阵文件合并或者去批次整合为一个基因表达矩阵,进行聚类并统计了每个分组中各个分群的细胞数的比例以及每个分群中各个分组细胞数的比例。该步骤将输出以Seurat格式储存的rds文件,若未选择进行细胞注释,则该整合后的rds结果用于后续拟时序、细胞通讯分析。
2.1.3 整合结果的标志基因鉴定
使用Seurat将整合后的聚类结果进行标志基因鉴定,生成标志基因表格、聚类热图、气泡图和点阵热图。若未选择进行细胞注释,则生成的标志基因表格csv将用于后续富集分析。
2.1.4 细胞注释
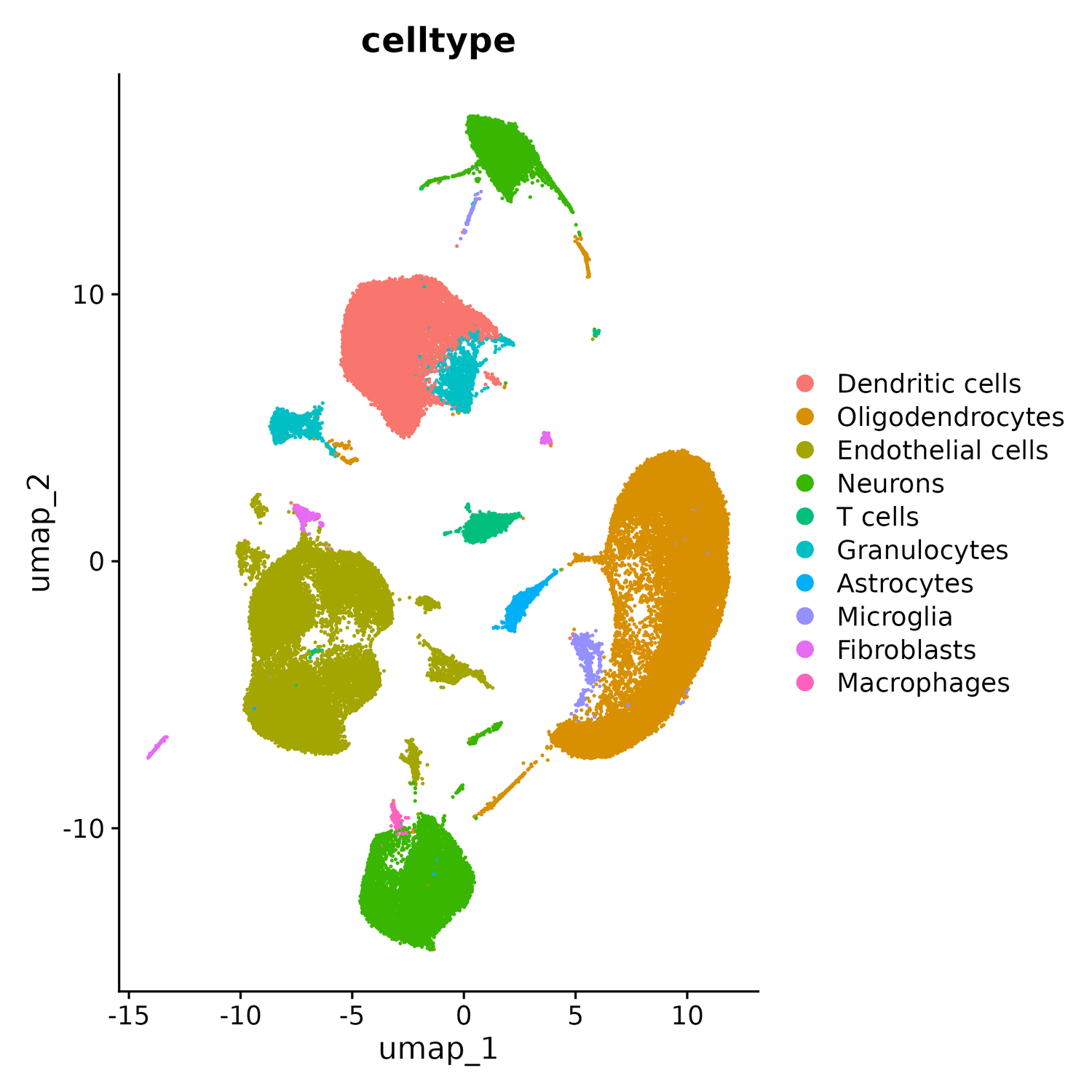
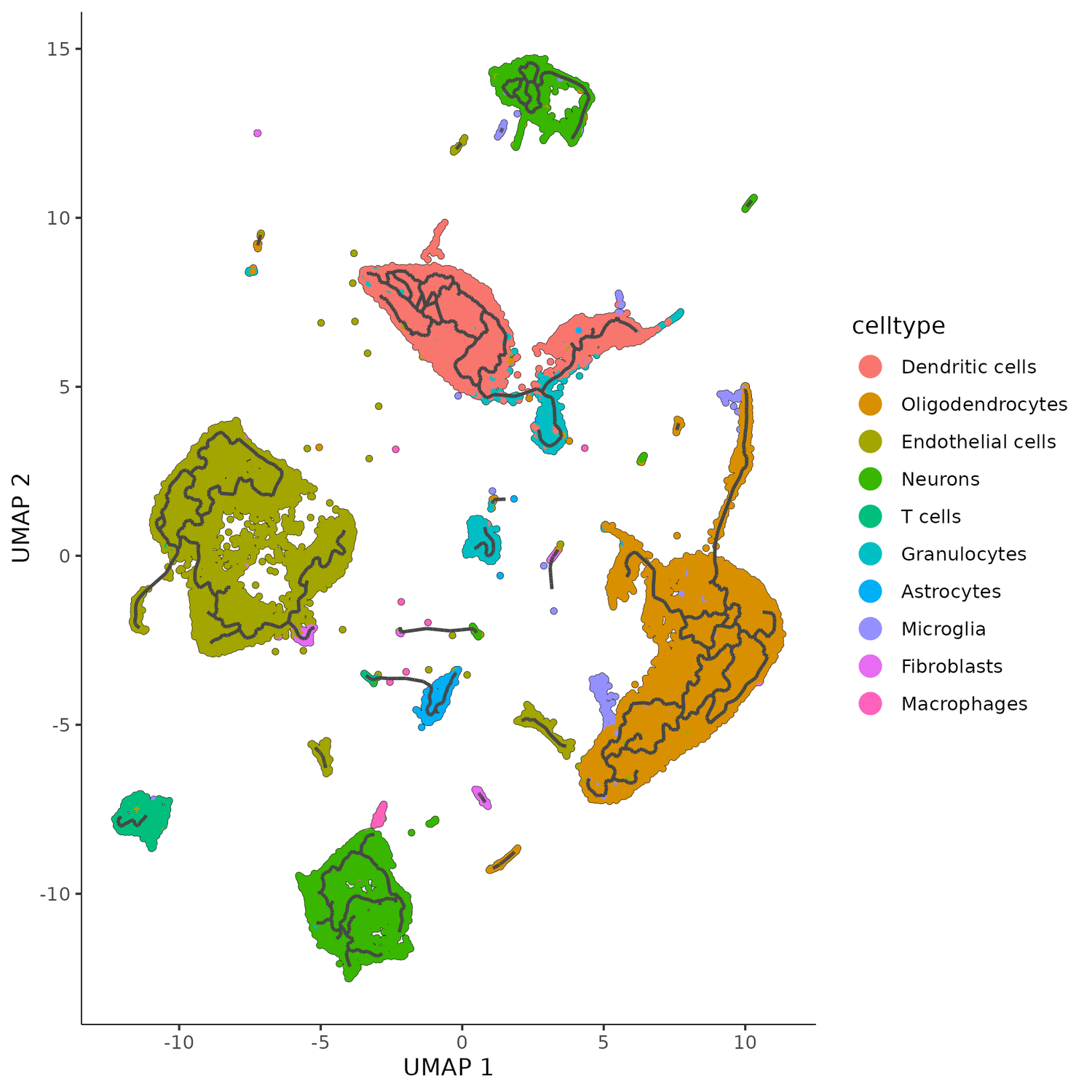
使用SingleR对整合后的结果进行细胞注释,将细胞聚类结果中的每个分群与参考数据库进行比较,根据该分群与参考数据库中已知细胞类群基因表达谱的相似性对注释结果进行打分,最终得到一个打分矩阵,选择最高得分为该分群的注释类型。该步骤将输出细胞类型注释图,生成细胞类型的标志基因表格并统计每个分组中各个细胞类型的细胞数的比例以及每个细胞类型中各个分组细胞数的比例。该步骤将输出以Seurat格式储存的rds文件,该rds文件和标志基因表格将会用于后续的拟时序、细胞通讯和富集分析。
2.1.5 拟时序分析
使用monocle3进行拟时序分析,基于Seurat对象提取矩阵进行降维聚类,并根据降维后的聚类结果之间的表达量关联以确定是否属于同一发展轨迹。该步骤输出细胞发育轨迹图、其中某个分群的拟时序图、某个分群的标志基因动态表达结果图和以monocle3格式储存的rds和tar文件,该结果可以使用monocl3::load_monocle_objects(‘结果文件路径’)来读取。
2.1.6 细胞通讯分析
使用CellChat进行细胞通讯分析,基于其内置的高质量信号分子互作数据库,对细胞进行差异表达分析和统计测试,推断不同细胞之间特定的通讯信号并可视化。该步骤输出细胞通讯网络图和以CellChat格式储存的rds文件。
2.1.7 富集分析
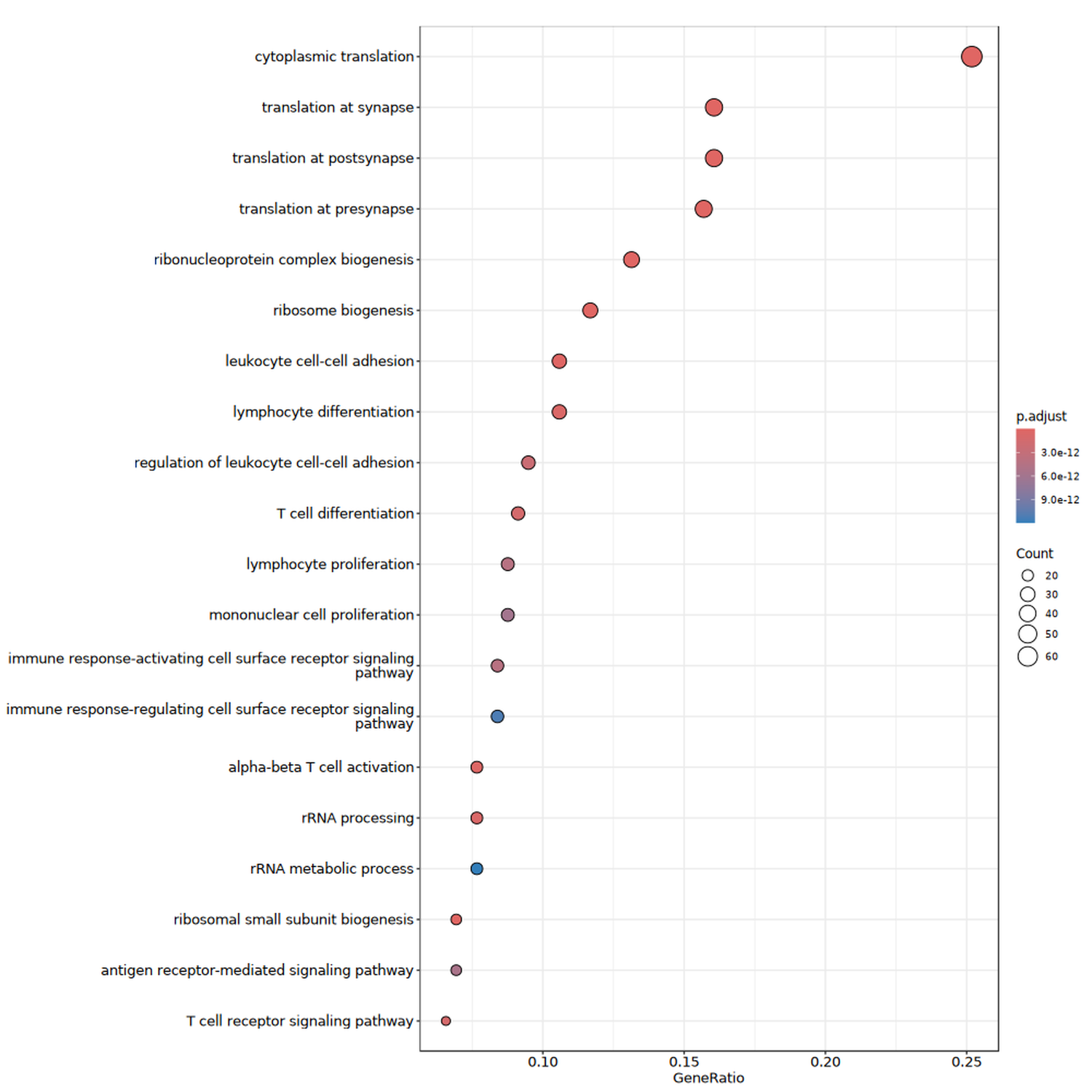
使用clusterProfiler对各个分群(或细胞类型)的标志基因进行KEGG通路分析和GO富集分析。
2.1.8 报告生成
综合分析结果,整理汇总成HTML报告。
第三章 使用说明书
Single-cell-multi-sample-anlysis标准分析流程通过DCS云平台系统进行样本输入及报告输出的全流程管理。下面具体介绍基于DCS云平台系统使用Single-cell-multi-sample-anlysis标准分析流程的操作指南。
3.1 指南概述
3.1.1 概述
本章介绍如何使用Single-cell-multi-sample-anlysis标准分析流程进行分析。在使用之前,请认真阅读并理解其中内容,保证能够正确使用。
3.2 使用场景一:手动投递
操作共包括四个步骤:上传数据、样本信息录入、启动分析。完成样本信息录入后,运行任务,当客户在看到任务状态为 completed 时,代表任务已完成,即可查看报告部分(详见3.4部分)。
3.2.1 步骤一:上传数据
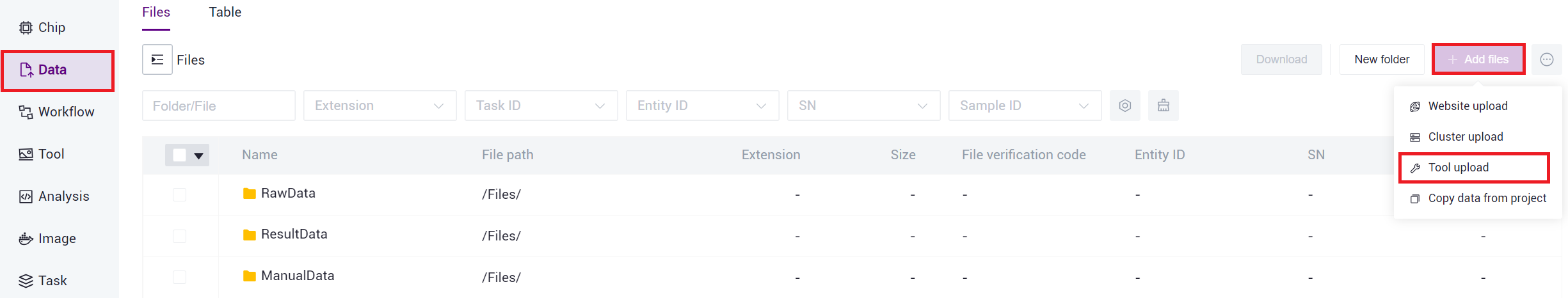
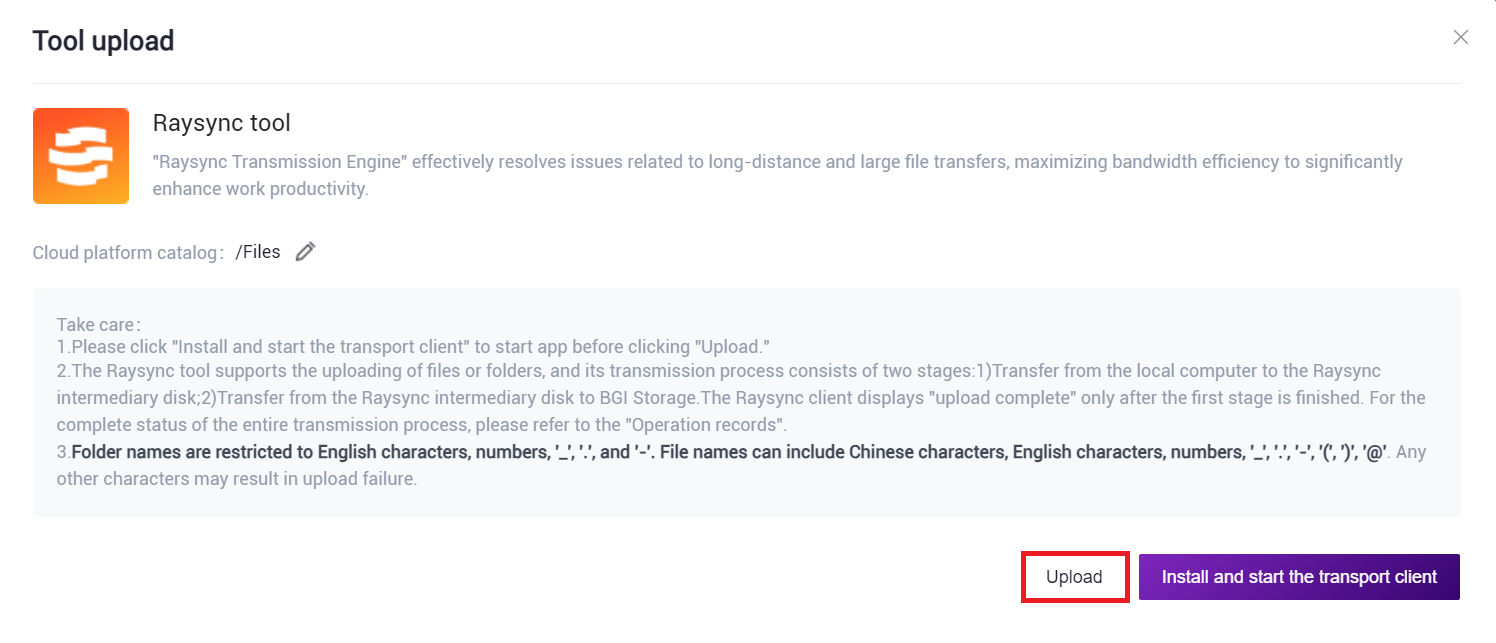
1)点击导航栏【Data】,进入数据管理页面,进入目标文件夹,点击右上角【+Add files】-【Tool upload】上传数据(图3-1):
- 点击【Upload】浏览并选择所需文件(图3-2),上传完成后文件会在目标文件夹中显示(如首次上传,则需点击【Install and start transport client】安装所需工具):
3.2.2 步骤三:样本信息录入
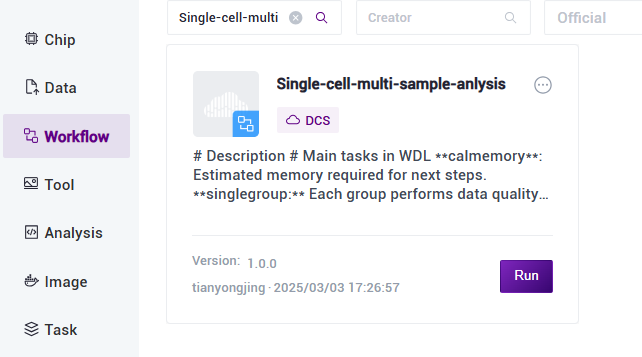
1)点击导航栏【Workflow】进入流程分析页面,在搜索框输入 Single-cell-multi-sample-anlysis,点击【Run】(图3-3):
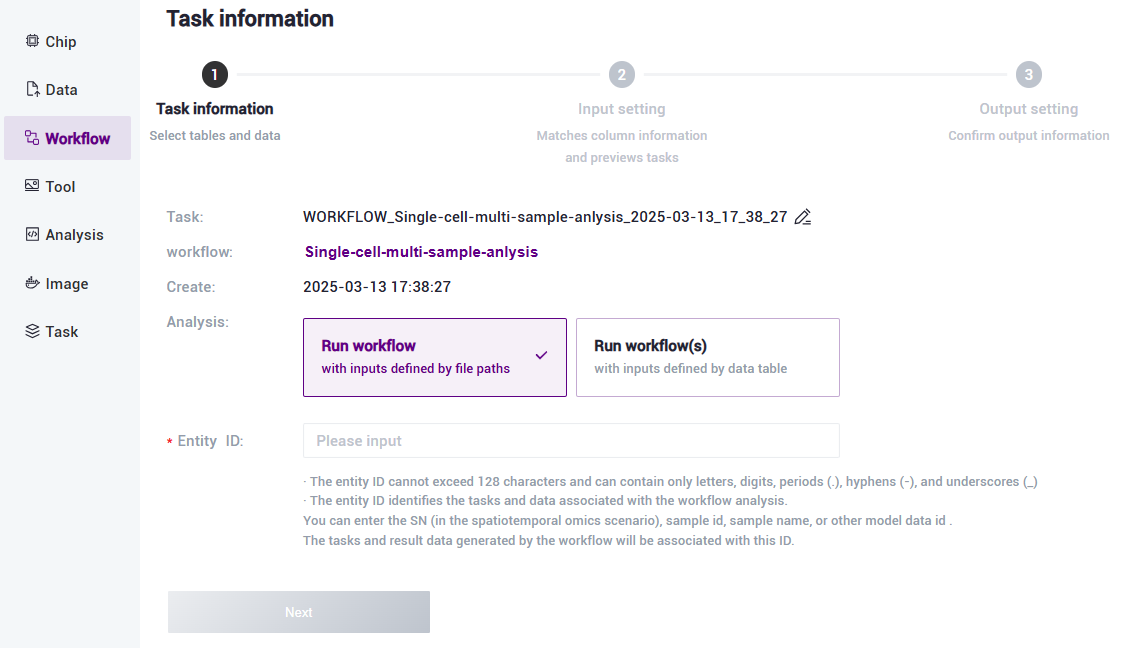
- 选择Run workflow,输入Entity ID,点击【Next】(图3-4):
- 录入样本信息,完成后点击【Next】(图3-5):
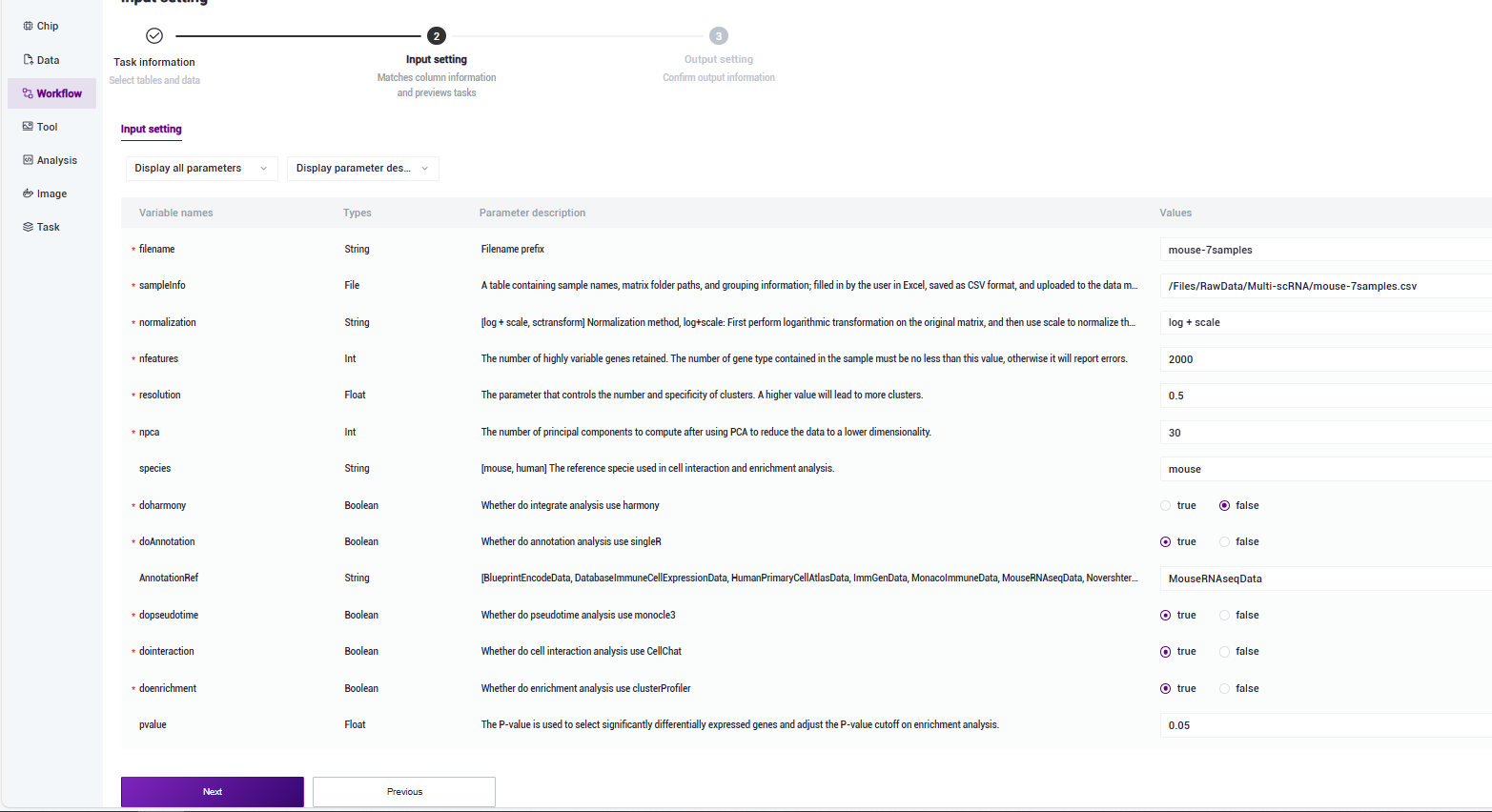
样本信息录入参数说明:
FileName:必填。输出文件名前缀;
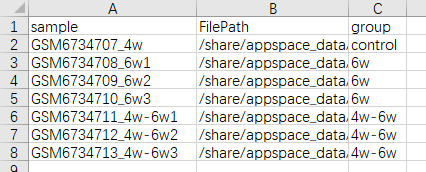
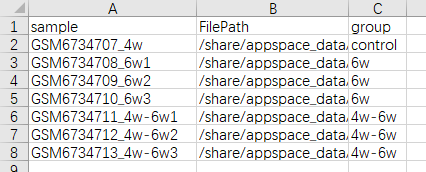
sampleInfo:必填。包含样本名(sample)、矩阵文件夹(FilePath)和分组信息(group)的表格; 由用户在excel中填写,保存为csv格式,上传至数据管理;
其中,样本名不可重复,由英文字母、数字和下划线组成;
样本测序文件路径要求是真实绝对路径,而非虚拟和相对路径,每个文件夹下包含3个文件barcodes.tsv.gz,features.tsv.gz和matrix.mtx.gz,文件名固定不可更改;
流程将自动判断features.tsv.gz格式,若超过2列判断为10X数据,读取第二列为gene_name,若仅一列判断为C4数据,读取第一列为gene_name;特殊格式暂无法正确判断gene_name位置。若gene_name位置存在问题,可能无法进行自动注释、细胞通讯分析和富集分析甚至会在聚类步骤报错。
DCS云平台真实绝对路径如下图:
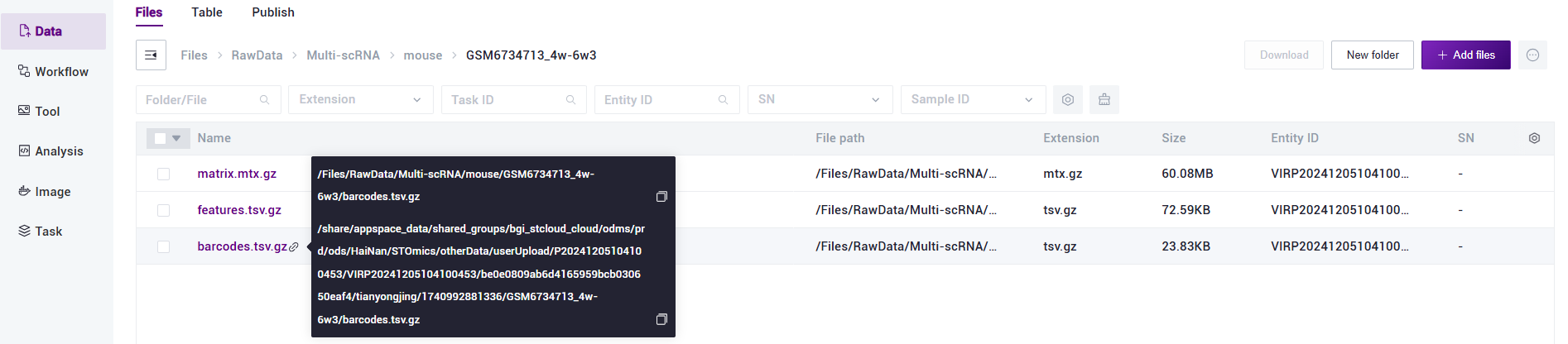
鼠标悬浮于文件旁边的🔗符号,复制红框中的真实文件夹路径(不要复制到具体文件名,复制到其上层文件夹,例如图中的文件夹复制到/……/GSM6734713_4w-6w3/这里即可),输入至表格中的FilePath
分组由英文字母、数字和下划线组成;
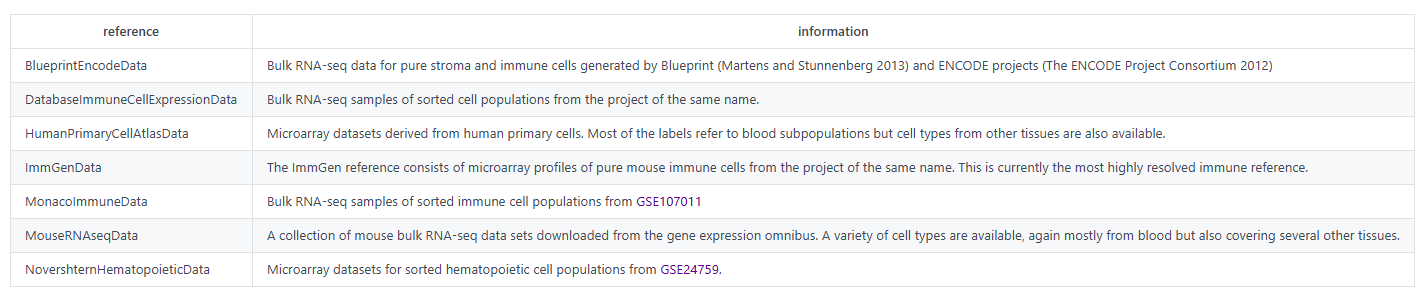
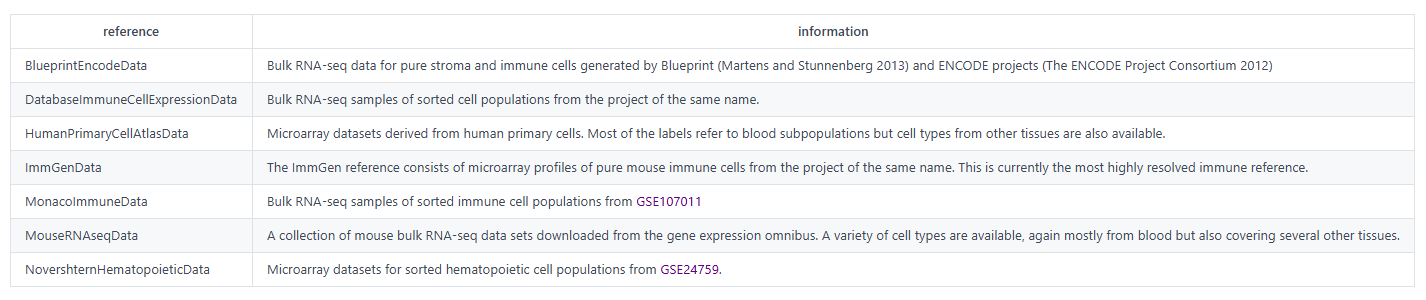
normalization:必填。对表达矩阵进行标准化的方法,可选参数为[log + scale, sctransform]。log + scale 是指使用Seurat中的NormalizeData和ScaleData进行标准化,而sctransform是指使用Seurat中的SCTransform进行标准化,两者算法差异可参见Seurat说明。 nfeatures:必填。保留高变异基因数,样本所含基因类型数必须不小于该值,否则会报错。默认保留2000个高变基因数。 resolution:必填。控制聚类数量和特异性的参数。值越高,聚类越多。默认为0.5 npca:必填。使用 PCA 将数据降低到较低维度后需要计算的主成分数量。默认为30 species:非必填。细胞通讯、富集分析所使用的参考物种,仅支持人或小鼠,只能填写human或者mouse,填写其他字符串均无法正常输出细胞通讯和富集分析的结果。 doharmony:必填。是否使用harmony对整合后的多样本结果进行去批次处理。默认不进行去批次处理。 doAnnotation:是否使用SingleR进行细胞类型注释。默认进行细胞注释。 AnnotationRef:非必填。若选择进行细胞注释时,则必须选择一个内置参考数据集进行细胞注释,可选参数为[BlueprintEncodeData, DatabaseImmuneCellExpressionData, HumanPrimaryCellAtlasData, ImmGenData, MonacoImmuneData, MouseRNAseqData, NovershternHematopoieticData] 。具体说明如下表:
dopseudotime:必填。是否使用monocle3进行拟时序分析。默认进行拟时序分析。 dointeraction:必填。是否使用CellChat进行细胞通讯分析。默认进行细胞通讯分析。 doenrichment:必填。是否使用clusterProfiler进行富集分析。默认进行富集分析。 pvalue:非必填。但是选择进行富集分析时,则必须设置P值。该参数用于筛选出差异表达显著的基因,并调整富集分析的P值截止值。
3.2.3 步骤四:启动分析
点击【Run】启动分析(图3-6):
3.3 使用场景二:表格投递
操作共包括五个步骤:上传数据、下载样本模板、填写并导入样本模板、启动分析。完成样本模板导入后,可批量运行任务,当客户在看到任务状态为completed 时,代表任务已完成,即可查看报告部分(详见3.4部分)
3.3.1 步骤一:上传数据
与使用场景一(手动投递)步骤一致(详见3.2.1步骤一:上传数据)
3.3.2 步骤三:样本信息录入表格下载
1) 点击导航栏【Data】,选择【Table】-【Download】(如图3-7), 点击【Workflow template】,选择 Single-cell-multi-sample-anlysis 模板下载:
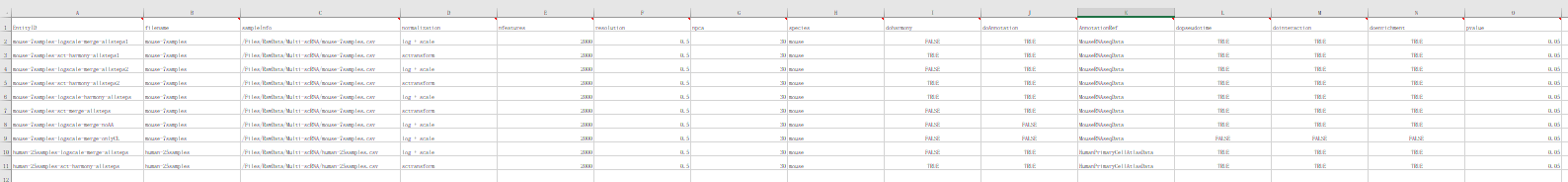
2)打开后Single-cell-multi-sample-anlysis样本模板Excel如图3-8:

3.3.3 步骤四:样本信息导入
1) 该使用场景条件下,样本导入表格需填写工作表(图3-15)。该场景表示导入表格后,直接进入分析。
Excel注意事项:
[1] 导入文件路径必须在云平台已存在,若不存在文件,则不会生成任务。
[2] 导入数据中,必填项字段不得为空。
[3] Excel中的EntityID需唯一。
[4] Excel中不能合并单元格,单元格内容前后不能有空格或特殊字符。
[5] 分析样本录入(图3-9):
FileName:必填。输出文件名前缀;
sampleInfo:必填。包含样本名(sample)、矩阵文件夹(FilePath)和分组信息(group)的表格; 由用户在excel中填写,保存为csv格式,上传至数据管理。
其中,样本名不可重复,由英文字母、数字和下划线组成。 样本测序文件路径要求是真实绝对路径,而非虚拟和相对路径,每个文件夹下包含3个文件barcodes.tsv.gz,features.tsv.gz和matrix.mtx.gz,文件名固定不可更改;
流程将自动判断features.tsv.gz格式,若超过2列判断为10X数据,读取第二列为gene_name,若仅一列判断为C4数据,读取第一列为gene_name;特殊格式暂无法正确判断gene_name位置。若gene_name位置存在问题,可能无法进行自动注释、细胞通讯分析和富集分析甚至会在聚类步骤报错。
DCS云平台真实绝对路径如下图:
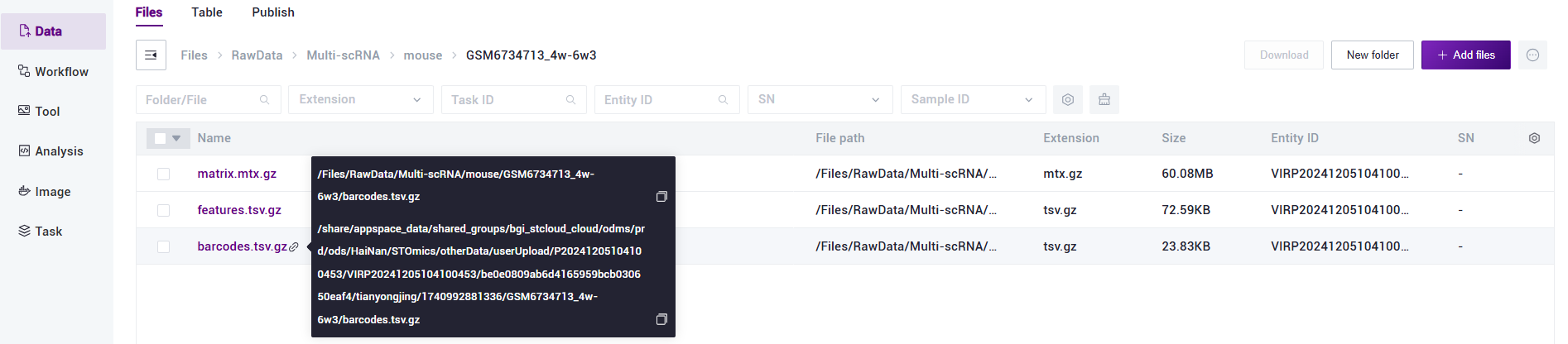
鼠标悬浮于文件旁边的🔗符号,复制红框中的真实文件夹路径(不要复制到具体文件名,复制到其上层文件夹,例如红框文件夹路径复制到/……/GSM6734713_4w-6w3/),输入至表格中的FilePath。
分组由英文字母、数字和下划线组成;
normalization:必填。对表达矩阵进行标准化的方法,可选参数为[log + scale, sctransform]。log + scale 是指使用Seurat中的NormalizeData和ScaleData进行标准化,而sctransform是指使用Seurat中的SCTransform进行标准化,两者算法差异可参见Seurat说明。
nfeatures:必填。保留高变异基因数,样本所含基因类型数必须不小于该值,否则会报错。可填写默认值2000。
resolution:必填。控制聚类数量和特异性的参数。值越高,聚类越多。可填写默认值0.5。
npca:必填。使用 PCA 将数据降低到较低维度后需要计算的主成分数量。可填写默认值30。
species:非必填。细胞通讯、富集分析所使用的参数物种,仅支持人或小鼠,只能填写human或者mouse,填写其他字符串均无法正常输出细胞通讯和富集分析的结果。
doharmony:必填。是否使用harmony对整合后的多样本结果进行去批次处理。可填写默认false,表示不进行去批次处理。
doAnnotation:必填。是否使用SingleR进行细胞类型注释。可填写默认值true,表示进行细胞注释。
AnnotationRef:非必填。若选择进行细胞注释,则必须选择一个内置参考数据集进行细胞注释,可选参数为[BlueprintEncodeData, DatabaseImmuneCellExpressionData, HumanPrimaryCellAtlasData, ImmGenData, MonacoImmuneData, MouseRNAseqData, NovershternHematopoieticData] 。具体说明如下表:
dopseudotime:必填。是否使用monocle3进行拟时序分析。可填写默认值true,表示进行拟时序分析。
dointeraction:必填。是否使用CellChat进行细胞通讯分析。可填写默认值true,表示进行细胞通讯分析。
doenrichment:必填。是否使用clusterProfiler进行富集分析。可填写默认值true,表示进行富集分析。
pvalue:非必填。但是选择进行富集分析时,则必须设置P值。该参数用于筛选出差异表达显著的基因,并调整富集分析的P值截止值。可填写默认值0.05。
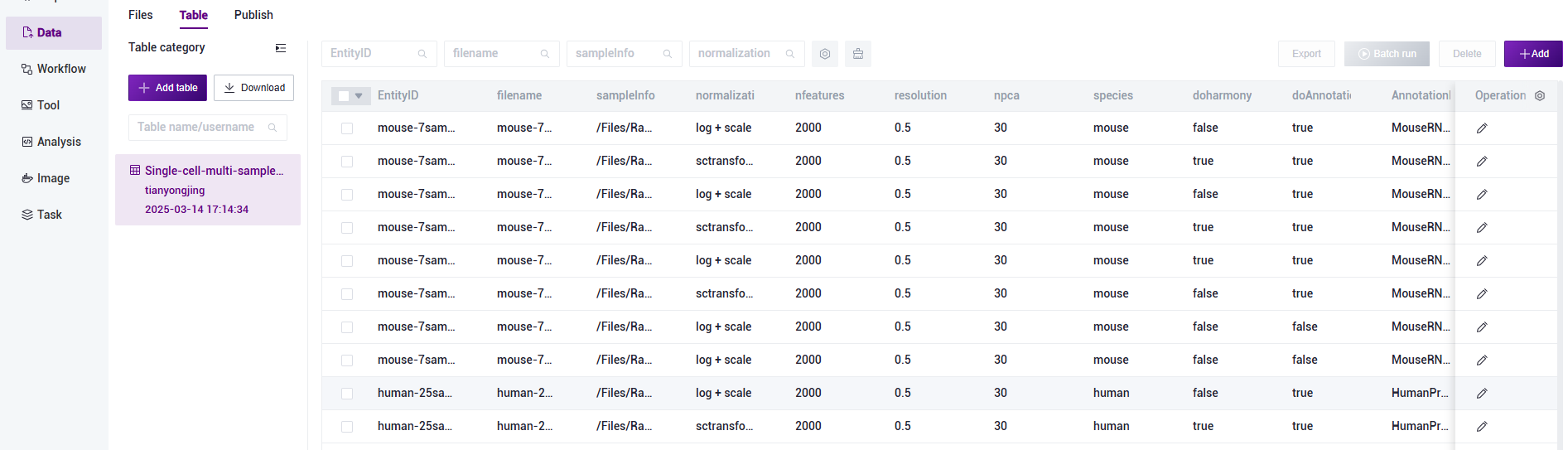
2) 配置好样本模板的分析样本录入工作表后,回到【Data】界面,点击【Table】-【+Add table】。点击【Click to upload/ Drop here】浏览并选择已填好样本信息的表格,点击【confirm】(图3-10):

3) 上传完成后文件会在目标文件夹中显示(图3-11):
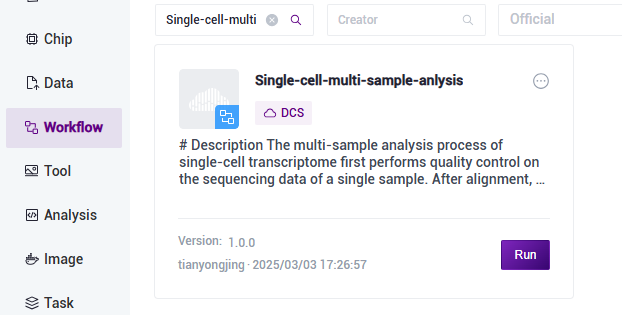
4) 点击导航栏【Workflow】进入流程分析页面,在搜索框输入Single-cell-multi-sample-anlysis,点击【Run】(图3-12):
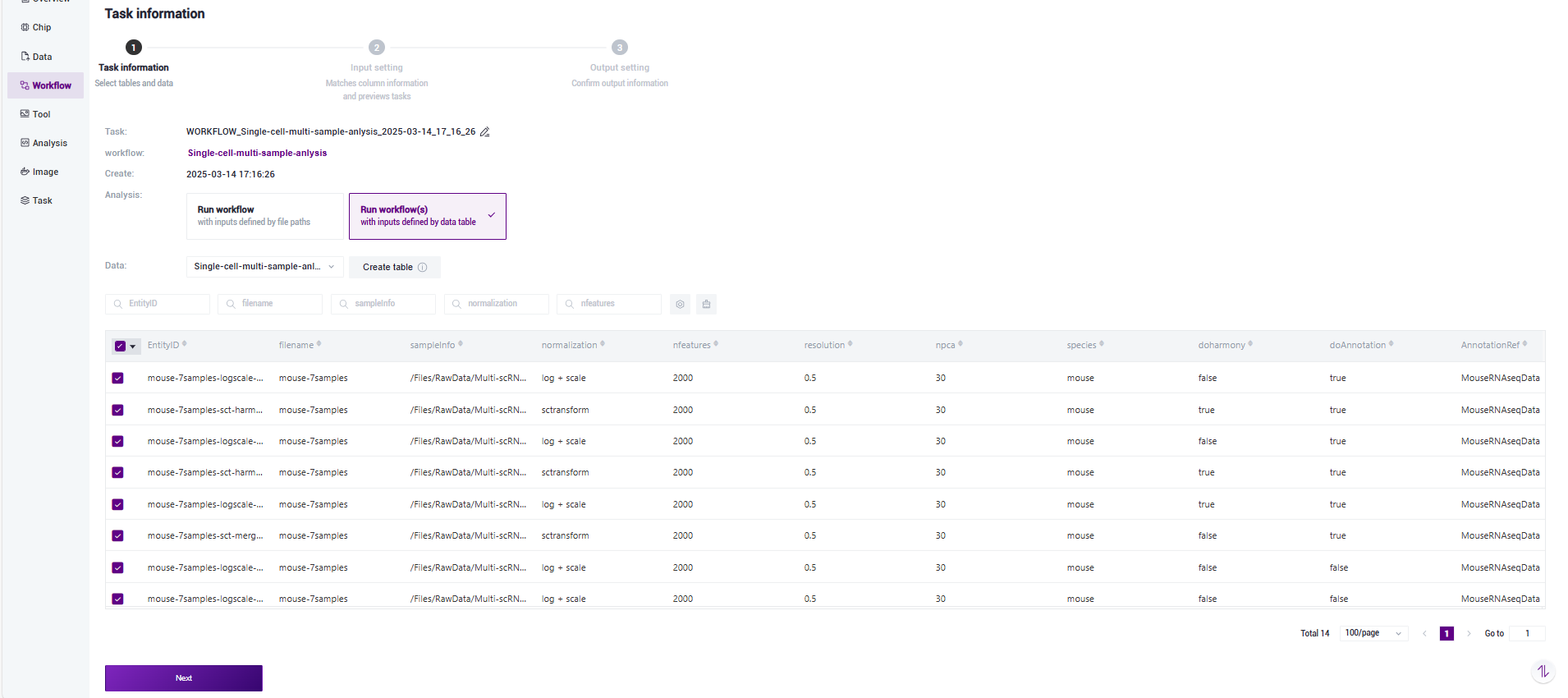
5) 选择Run workflow(s),点击 Please select table 处,选择在本小节3)中导入的表格,选中所需的行,点击【Next】(图3-13):
6) Values一般会自动填充,仅需要检查填充是否正确,若需修改则在Values处点击 并选择对应的值(如图3-14):
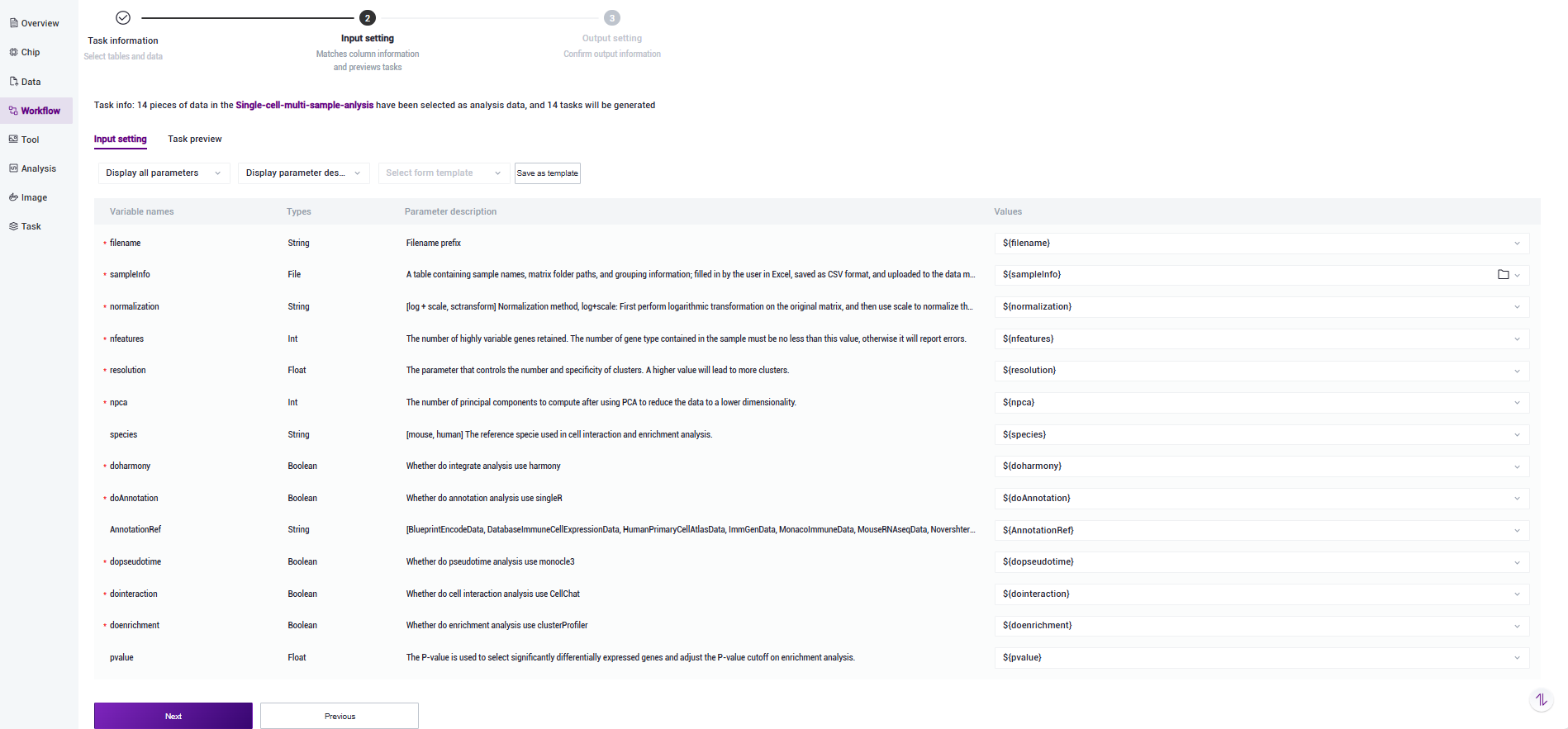
3.3.4 步骤五:启动分析
点击【Run】启动分析(图3-15):
3.4 报告查看及结果文件下载
3.4.1 报告查看
点击导航栏【Task】,当任务状态显示为 completed时,代表任务已完成,点击【Detail】即可查看结果部分(如图3-16):
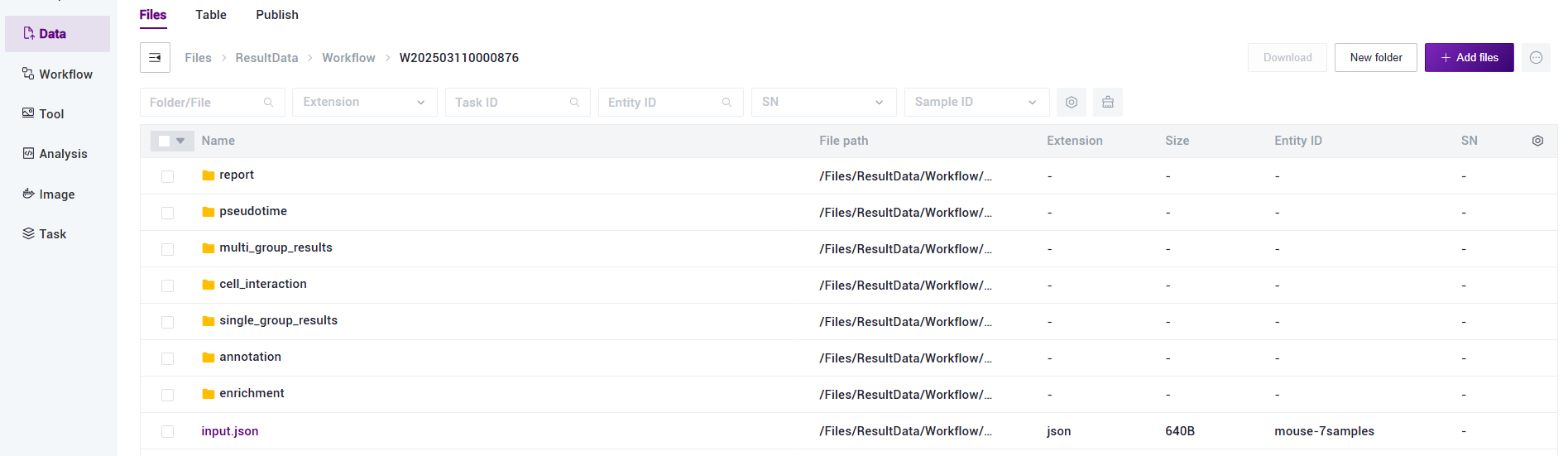
3.4.2 报告下载
- 点击导航栏【Data】,进入数据管理页面,根据任务的Task ID进行搜索,点击进入Task ID文件夹(如图3-17):
- 点击进入 report文件夹(如图3-18):
- 选中 *report.html 文件,点击【Download】-【Web download】(如图3-19):
第四章 结果展示
4.1 Single-cell-multi-sample-anlysis样本报告展示