LUSH_Germline_FASTQ_WGS 用户手册
第一章 产品信息
1.1 产品描述
LUSH流程是基于 GATK 最佳实践的优化流程。其主要组成部分是DCS Tools的aligner、 bqsr、 variantCaller、 genotyper 和 report。该流程功能包括数据比对、质控、变异检测。通过DCS Cloud平台用户图形化界面进行参数输入及结果输出的流程分析管理,使用方法简单。该流程使用高性能服务器配置,任务实行并行化,缩短分析周期,达到快速、高效交付的目的。
1.2 注意事项
- 本产品仅用于科研用途,不用于临床诊断,使用前请仔细阅读本说明书。
- 本说明书及其包含的信息为深圳华大生命科学研究院(以下简称深圳华大研究院)的专有保密信息,未经深圳华大研究院的书面许可,任何个人或组织不得全部或部分地对本说明书进行重印、复制、修改、传播或公布给他人。本说明书的读者为终端用户,由深圳华大研究院授权终端用户予以使用。严禁未授权的个人使用本说明书。
- 深圳华大研究院对本说明书不做任何种类的保证,包括(但不限于)用于特定目的的商业性和合理性的隐含保证。深圳华大研究院已经采取措施,确保本说明书的准确性。但是,深圳华大研究院对错误或遗漏不承担责任,并保留任何对本说明书和流程进行改进以提高其可靠性、功能或设计的权利。
- 本说明书中的所有图片均为示意图,图片内容可能与使用界面有细微差异,请以实际使用界面为准。
第二章 产品介绍
2.1 分析流程图
LUSH流程是基于 GATK 最佳实践的优化流程。其主要组成部分是DCS Tools的aligner、 bqsr、 variantCaller、 genotyper 和 report。该流程功能包括数据比对、质控、变异检测。通过DCS Cloud平台用户图形化界面进行参数输入及结果输出的流程分析管理,使用方法简单。该流程使用高性能服务器配置,任务实行并行化,缩短分析周期,达到快速、高效交付的目的。
流程包含以下几项功能:
数据比对:根据参考基因组进行比对,生成并统计比对结果。
变异检测:根据比对结果,分析样本的突变结果包括SNP和INDEL。
图2-1是分析整体流程图:
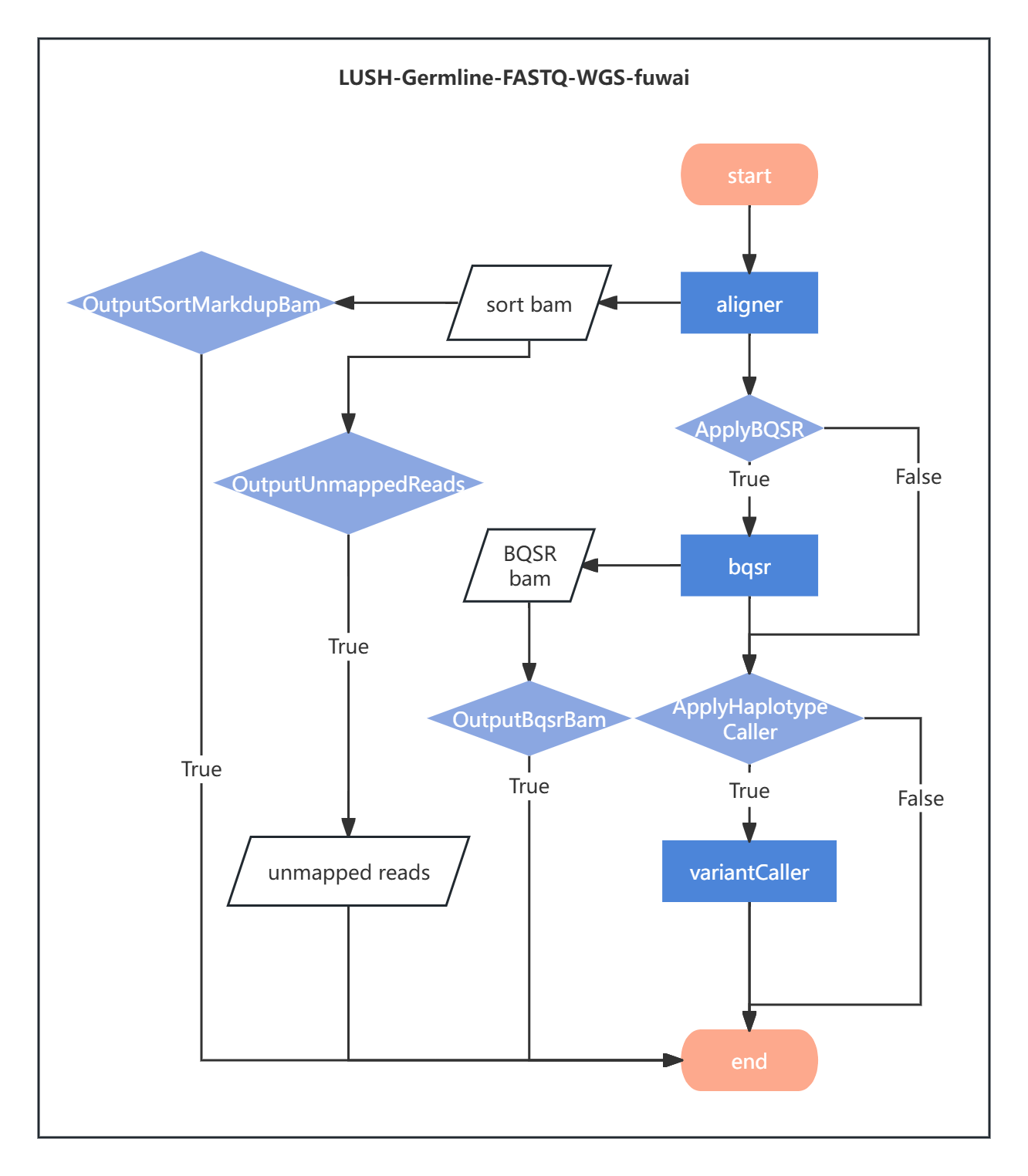
2.1.1 参考基因组过滤、比对、排序与去重
将序列比对到参考基因组上确定每条序列的位置,为进一步检测变异提供基础;标记重复消除PCR扩增产生的重复序列,降低假阳性提高变异检测准确性,这些步骤由软件alinger完成。而且aligner 还集成了数据质控功能,可以在序列比对之前对序列进行过滤和处理,从而降低错误率,避免后续分析受到噪音数据的干扰。
2.1.2 碱基质量矫正
由于存在系统误差,序列碱基质量值并不总是完全准确,需要对序列的碱基质量值重校正。该步骤可以纠正系统偏差生成更精确的质量分数,提高变异检测准确性,减少假阳性和假阴性。统一质量评估标准符合GATK推荐的最佳实践,该步骤由软件bqsr完成。
2.1.3 变异检测
variantCaller 是 GATK HaplotypeCaller 的 C/C++ 重新实现,以及genotyper 是 GenotypeGVCFs 的 C/C++ 重新实现。使用variantCaller工具进行变异检测,生成GVCF文件。并genotyper工具生成Genotype VCF文件。
2.1.4 报告生成
report 负责整合前面任务输出的统计文件,生成图像和html报告。
第三章 使用说明书
LUSH标准分析流程通过DCS智能云平台进行样本输入及结果输出的全流程管理。下面具体介绍基于DCS智能云平台使用LUSH标准分析流程的操作指南。
3.1 指南概述
3.1.1 概述
本章介绍如何使用LUSH标准分析流程进行分析。在使用之前,请认真阅读并理解其中内容,保证能够正确使用LUSH。
3.1.2 流程套件
- LUSH_Germline_FASTQ_WGS_Human
- LUSH_reference_index
- LUSH_Germline_FASTQ_WGS_NHS
- ExpansionHunter_WGS_STR
- CNVpytor_WGS_CNV
- PanGenie_WGS_SV
3.2 使用场景-LUSH主流程:手动投递
操作共包括四个步骤:上传数据、添加工作流、LUSH2参考基因组索引构建、LUSH2分析。运行任务后,当客户在看到任务状态为![]() completed时,代表任务已完成。
completed时,代表任务已完成。
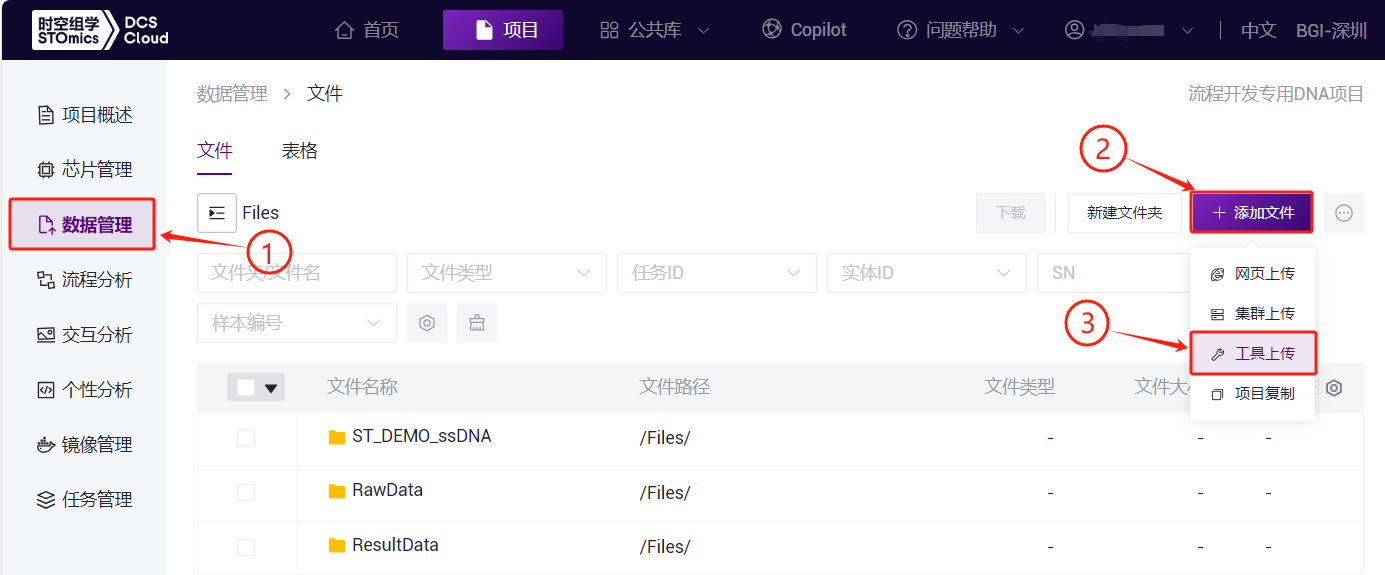
3.2.1 步骤一:上传数据
点击左侧导航栏【数据管理】,进入数据管理页面,进入目标文件夹,点击右上角【+添加文件】-【工具上传】上传数据(图3-1):
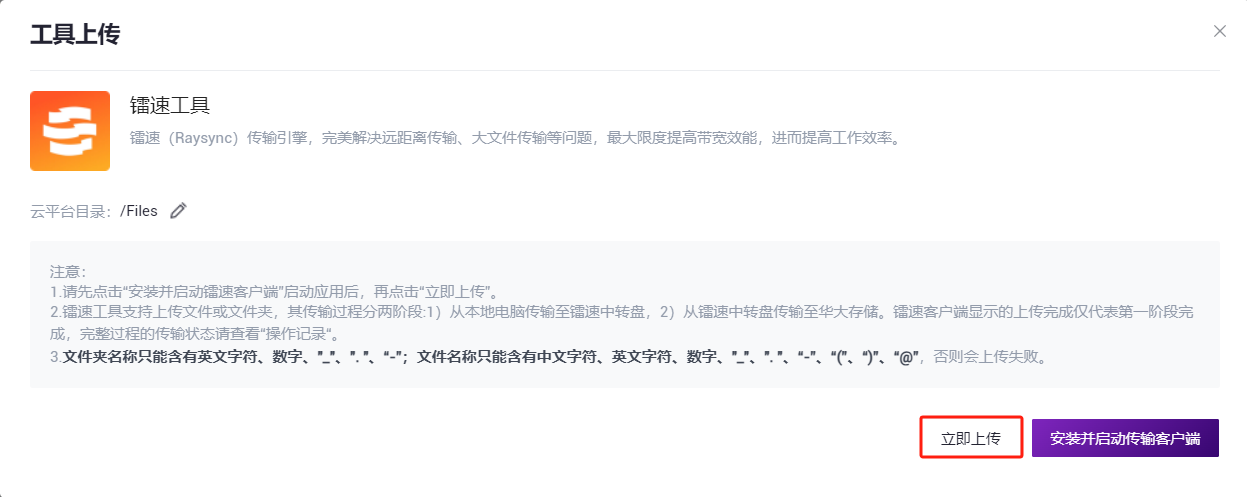
- 点击【上传】浏览并选择所需文件(图3-2),上传完成后文件会在目标文件夹中显示(如首次上传,则需点击【安装并启动传输客户端】安装所需工具):
3.2.2 步骤二:添加工作流
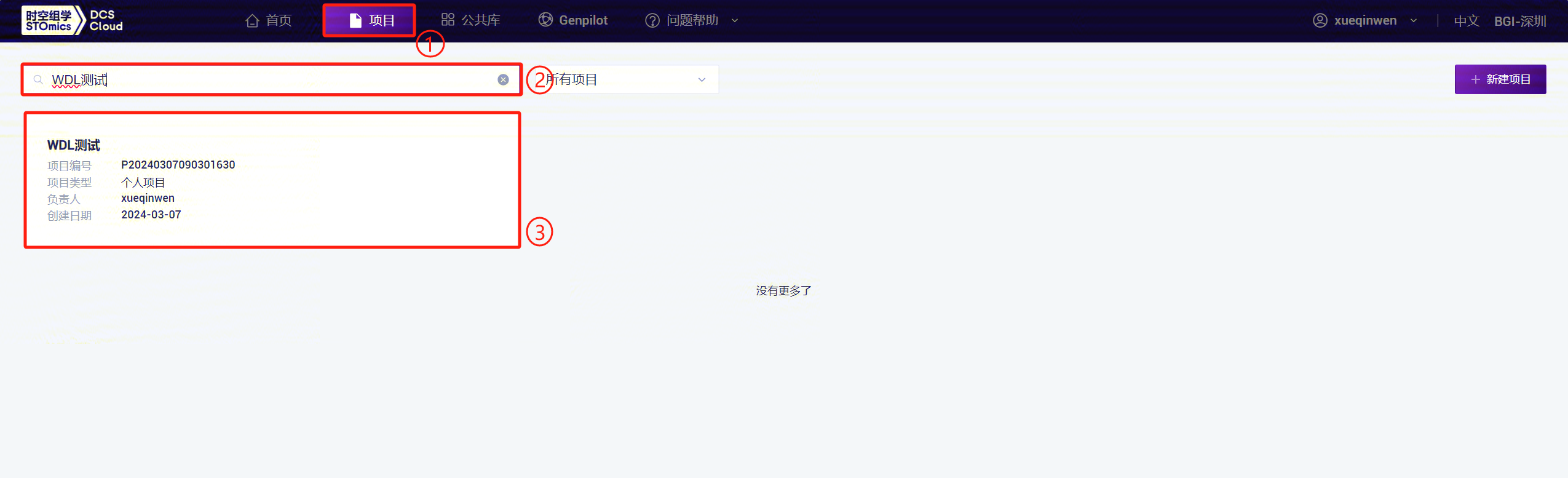
- 点击顶部导航栏【项目】进入项目列表页面,在搜索框输入项目名称/项目编号,点击进入项目(图3-3):
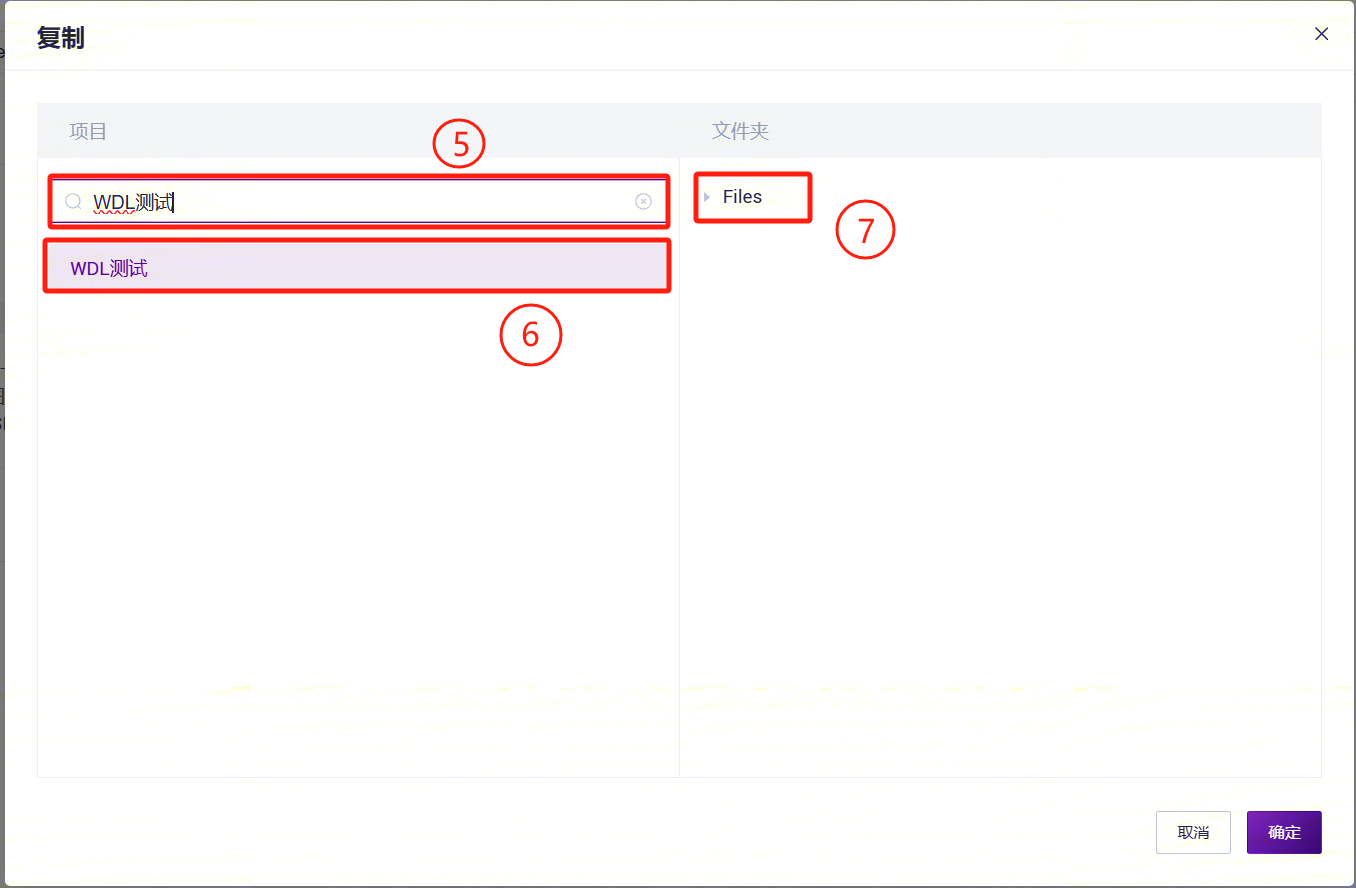
- 点击左侧导航栏【流程分析】进入流程分析页面,点击右上角【+ 添加工作流】添加工作流,点击【从公共库复制】(图3-4):
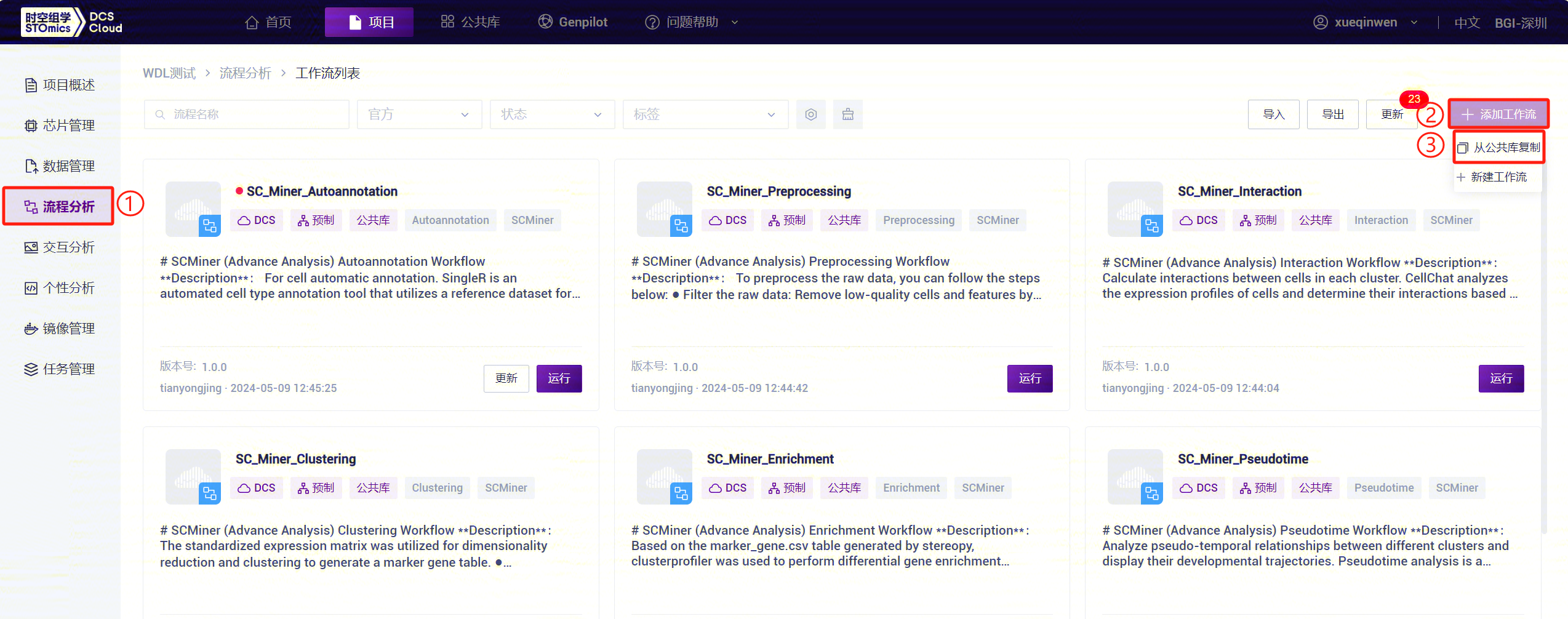
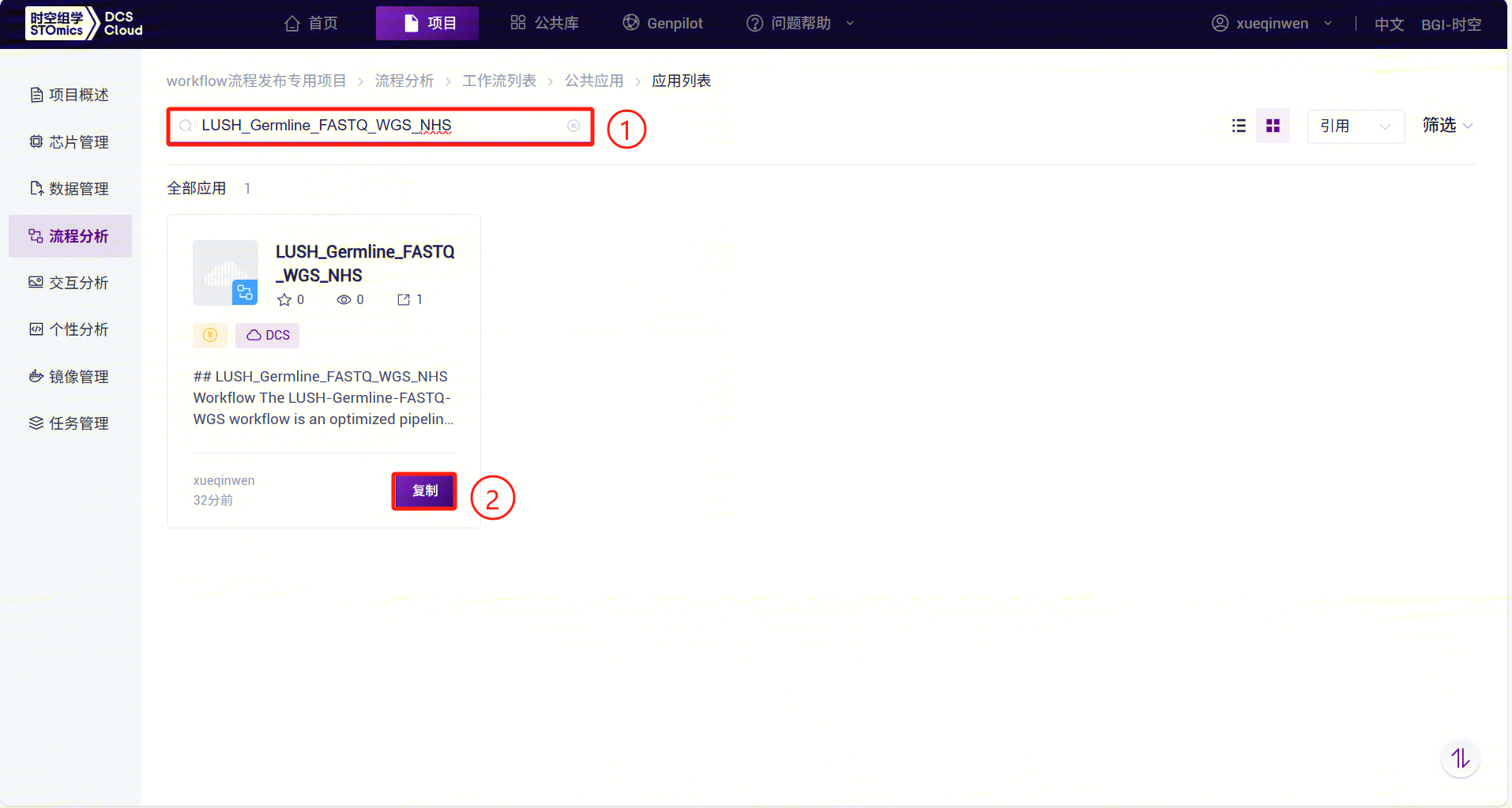
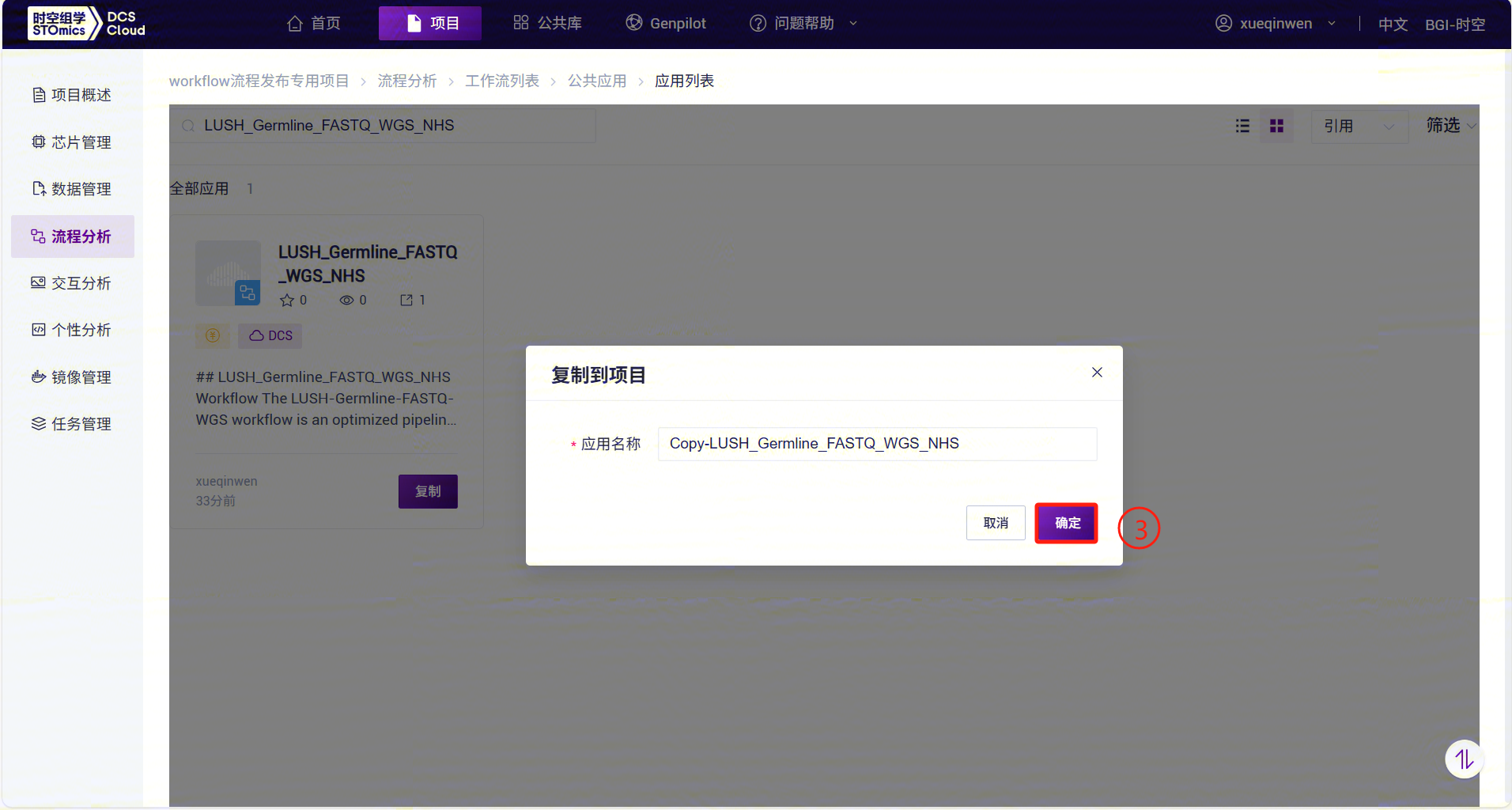
- 以LUSH_Germline_FASTQ_WGS_NHS为例,在搜索框中输入流程名称LUSH_Germline_FASTQ_WGS_NHS,选择, 点击【复制】,点击【确定】,其他流程套件,也进行相同的操作:
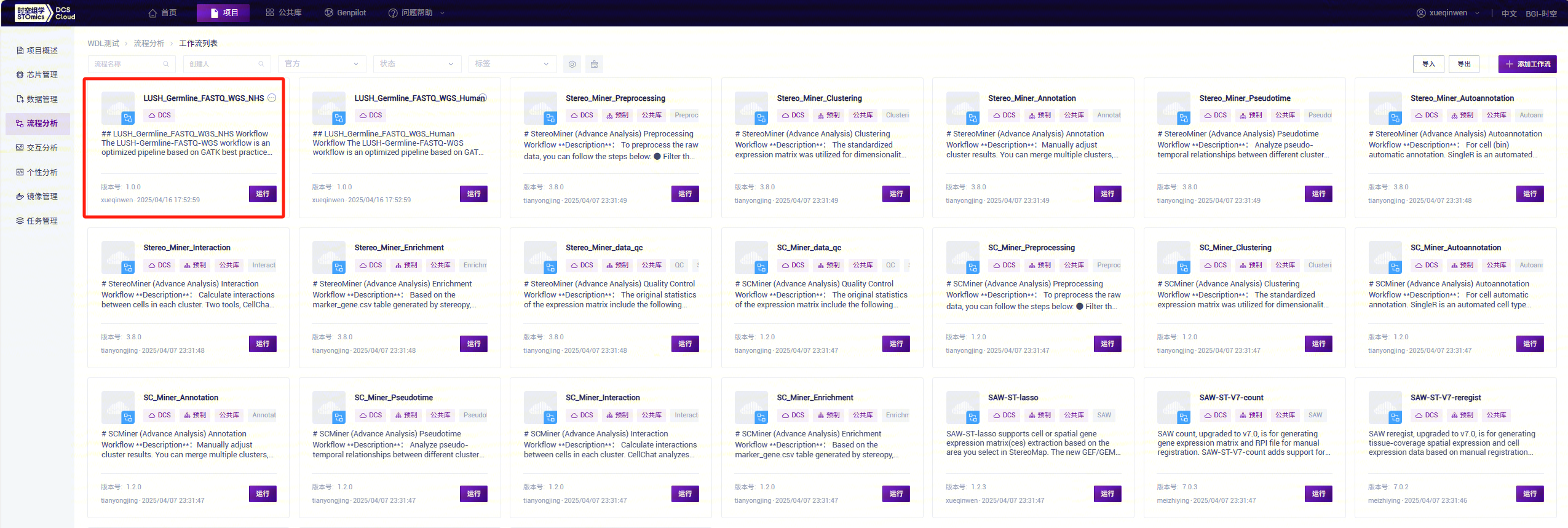
- 工作流添加成功:
3.2.3 步骤三:LUSH参考基因组索引构建
- 对于其他物种基因组分析,LUSH流程支持用户提供构建参考基因组索引。
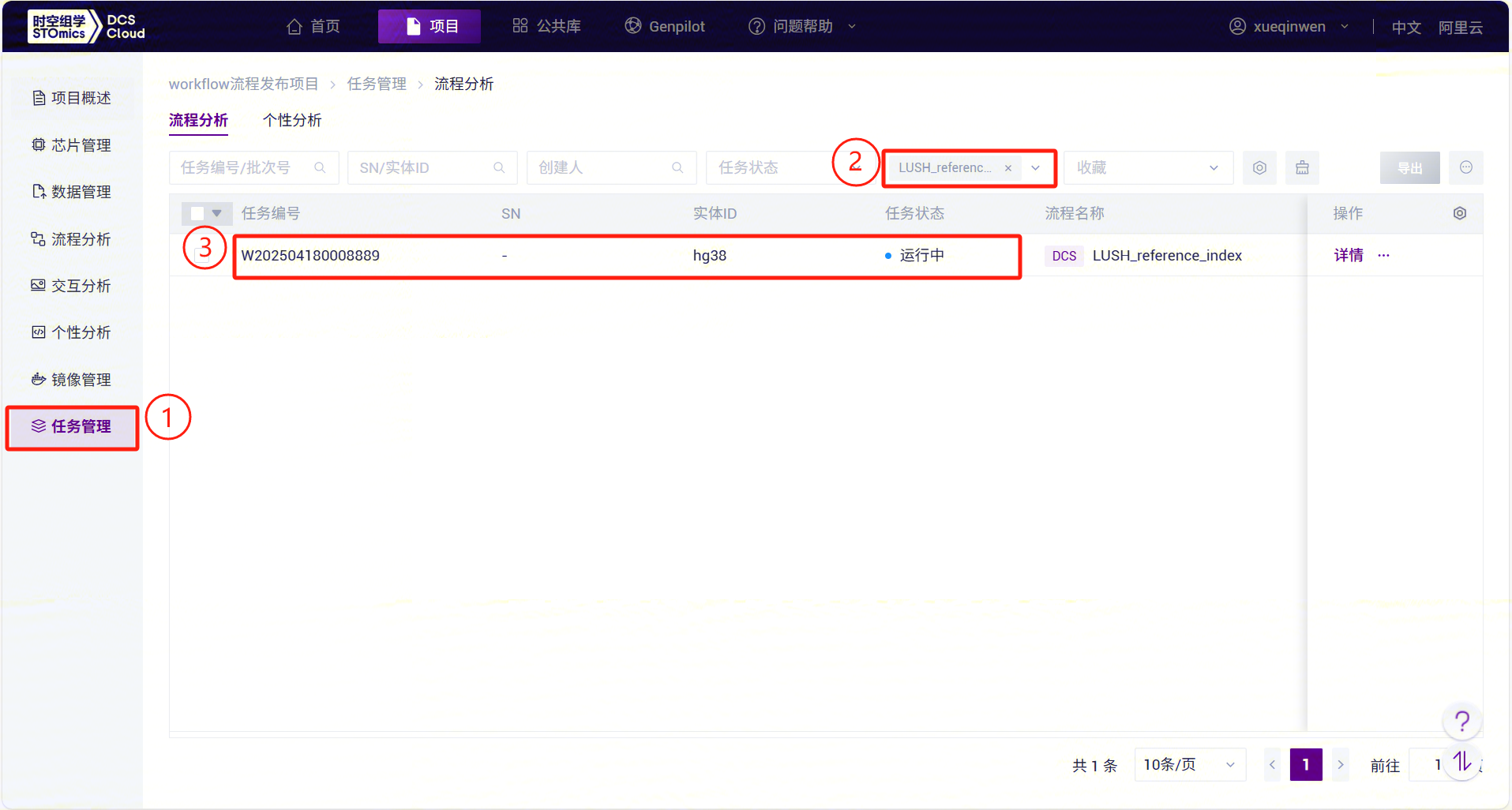
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入LUSH_reference_index,点击【运行】:
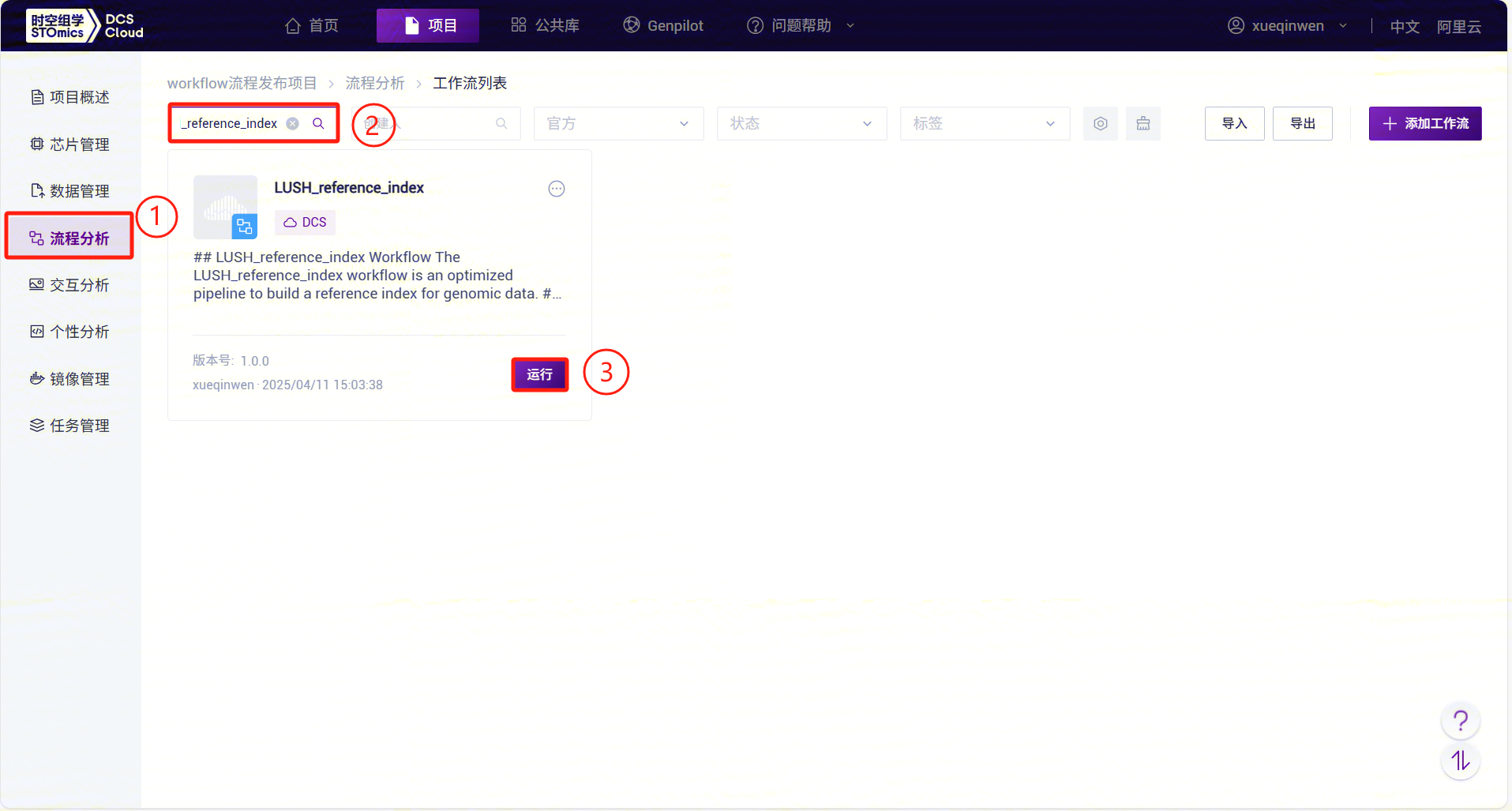
- 输入实体 ID,点击【下一步】(图3-8):
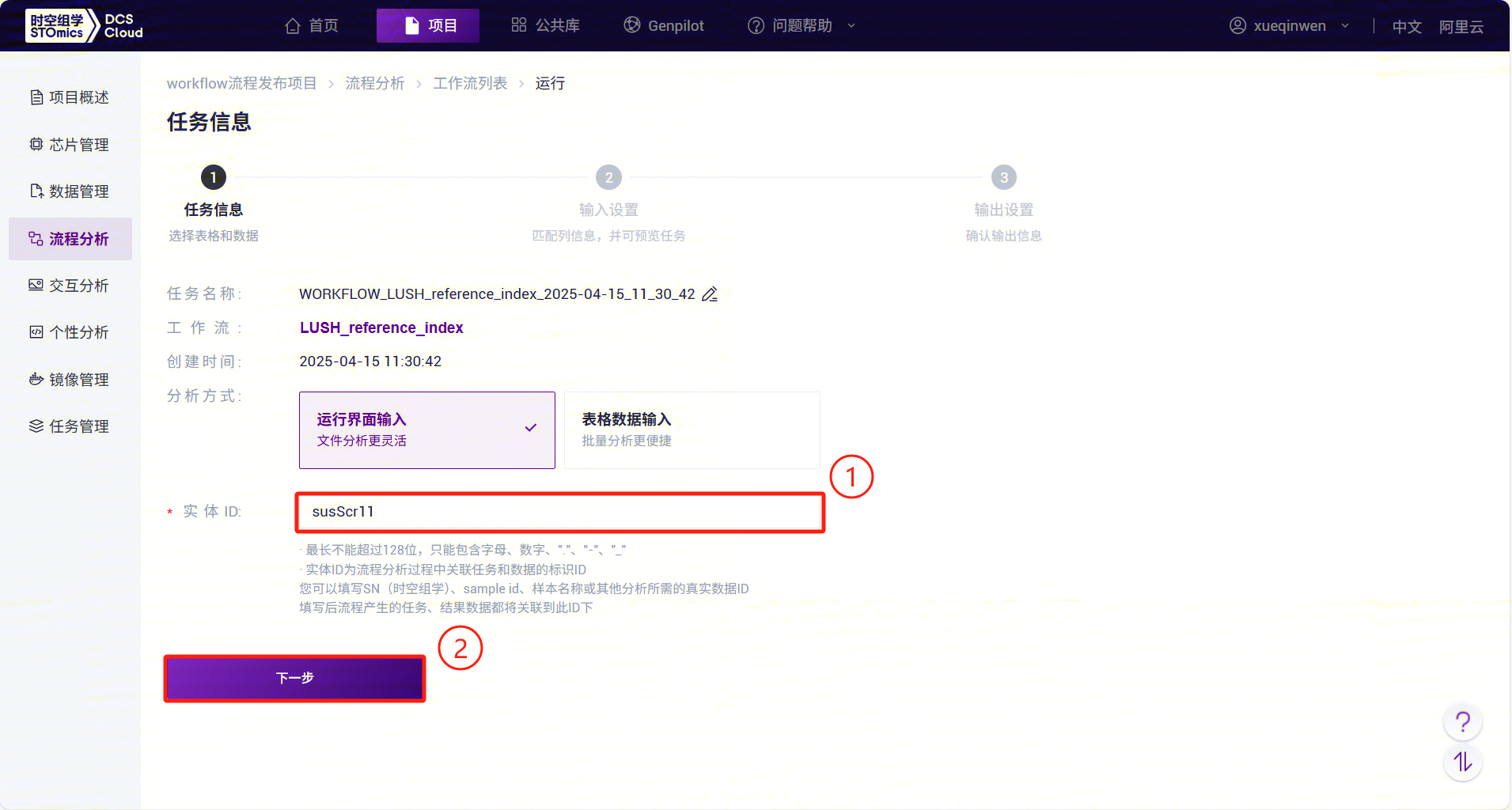
- LUSH录入参数信息,完成后点击【下一步】(图3-9):
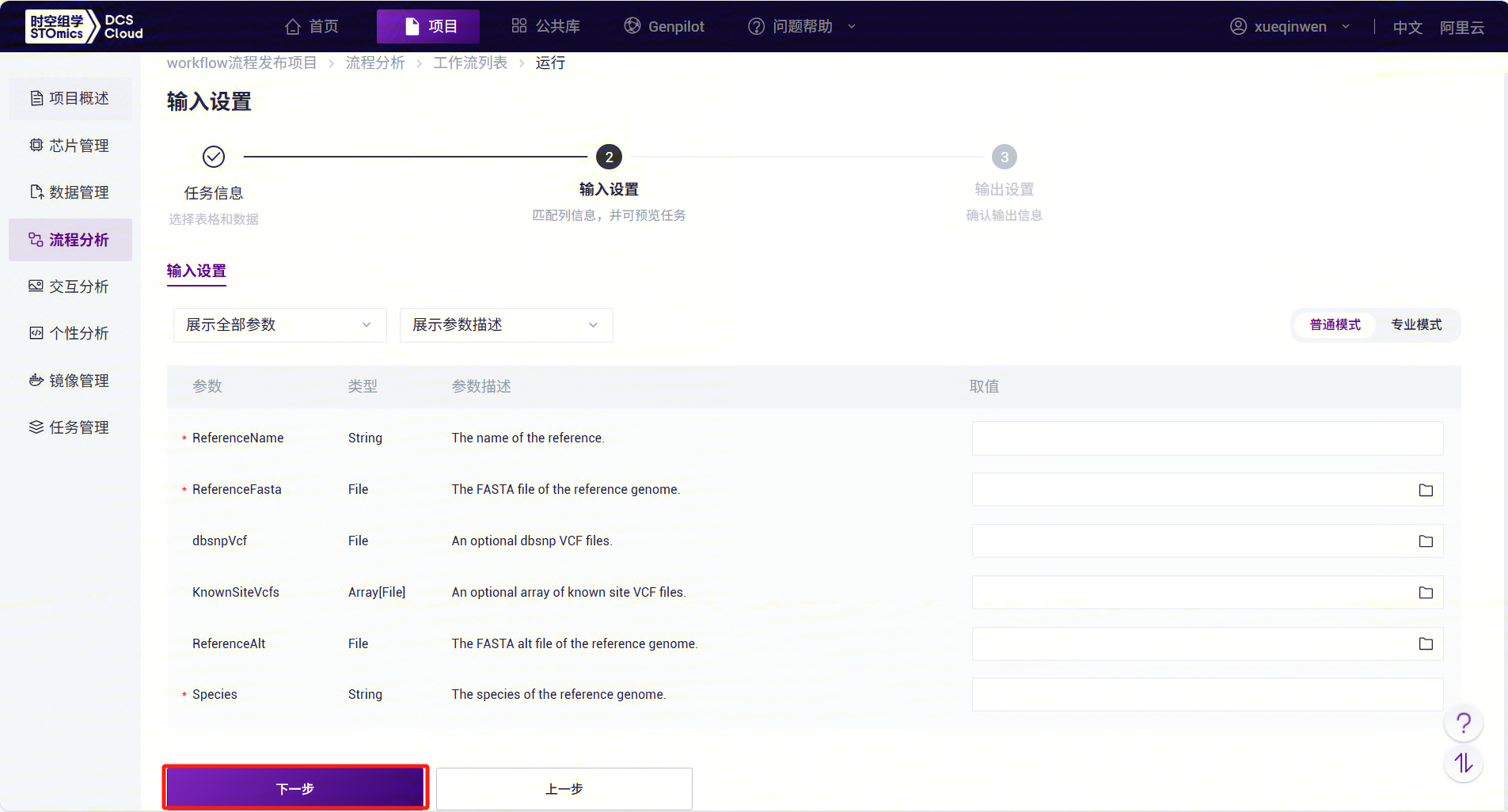
LUSH_build_index流程变量说明:
- ReferenceName:参考基因组名称;
- ReferenceFasta:参考基因组FASTA文件;
- dbsnpVcf:参考基因组的dbSNP数据库vcf文件(选填)。
- KnownSiteVcfs:包含该物种已知位点的vcf文件(选填);
- ReferenceAlt:包含该物种alt信息的索引文件(选填);
- Species:物种名。
- 点击【运行】启动分析(图3-10)
- 点击左侧导航栏【任务管理】进入流程分析页面,在流程名称中选择LUSH_build_index,查看任务状态,任务完成后,任务状态显示为
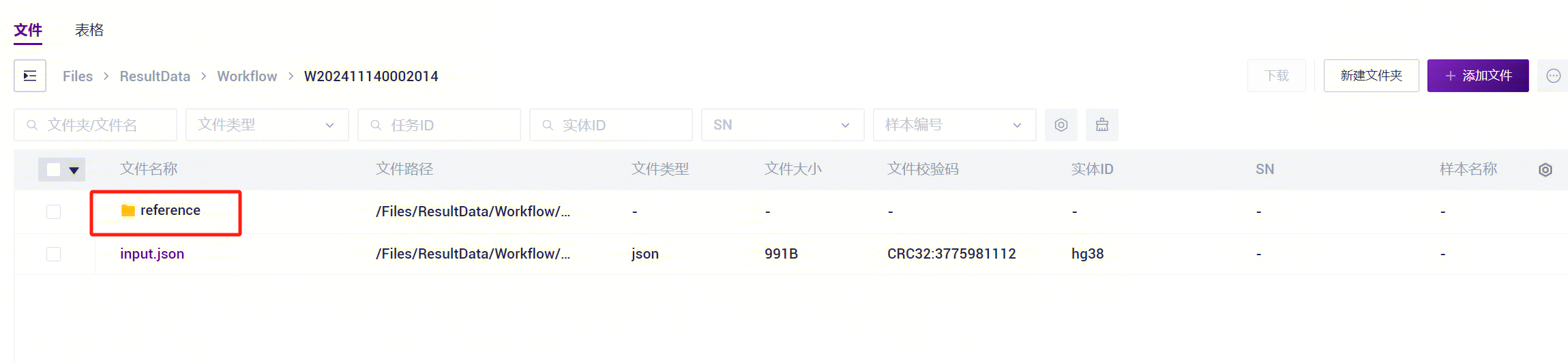
 完成,复制任务编号,点击导航栏【数据管理】,将任务编号输入文件夹/文件名进行搜索。进入结果文件,其中 reference 文件夹作为 LUSH_Germline_FASTQ_WGS_NHS 流程中 ReferenceDir 参数输入。
完成,复制任务编号,点击导航栏【数据管理】,将任务编号输入文件夹/文件名进行搜索。进入结果文件,其中 reference 文件夹作为 LUSH_Germline_FASTQ_WGS_NHS 流程中 ReferenceDir 参数输入。
3.2.4 步骤四:LUSH_Germline_FASTQ_WGS_NHS 分析流程
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入LUSH_Germline_FASTQ_WGS_NHS,点击【运行】(参考3.2.3):
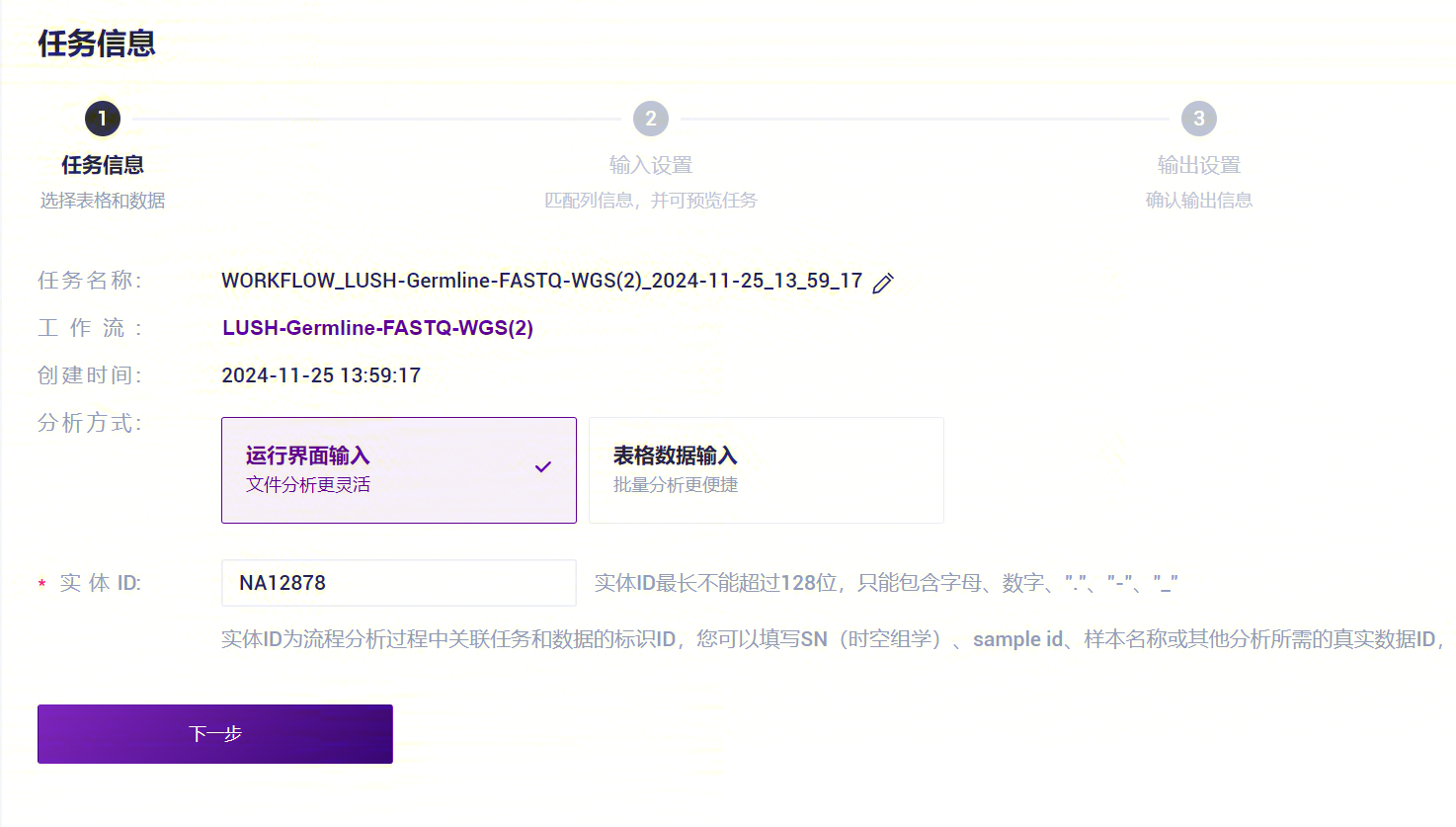
- 输入实体 ID(一般为样品ID),点击【下一步】(参考3.2.3):
- LUSH_Germline_FASTQ_WGS_NHS录入样本信息。PE FASTQ 输入:根据成对的 FASTQ 文件数量,点击“+”按钮添加文件组,并在每个文件组中选择对应的 FASTQ1 和 FASTQ2 文件; SE FASTQ输入:操作与 PE 数据类似,每个文件组中放置一个 FASTQ 文件。
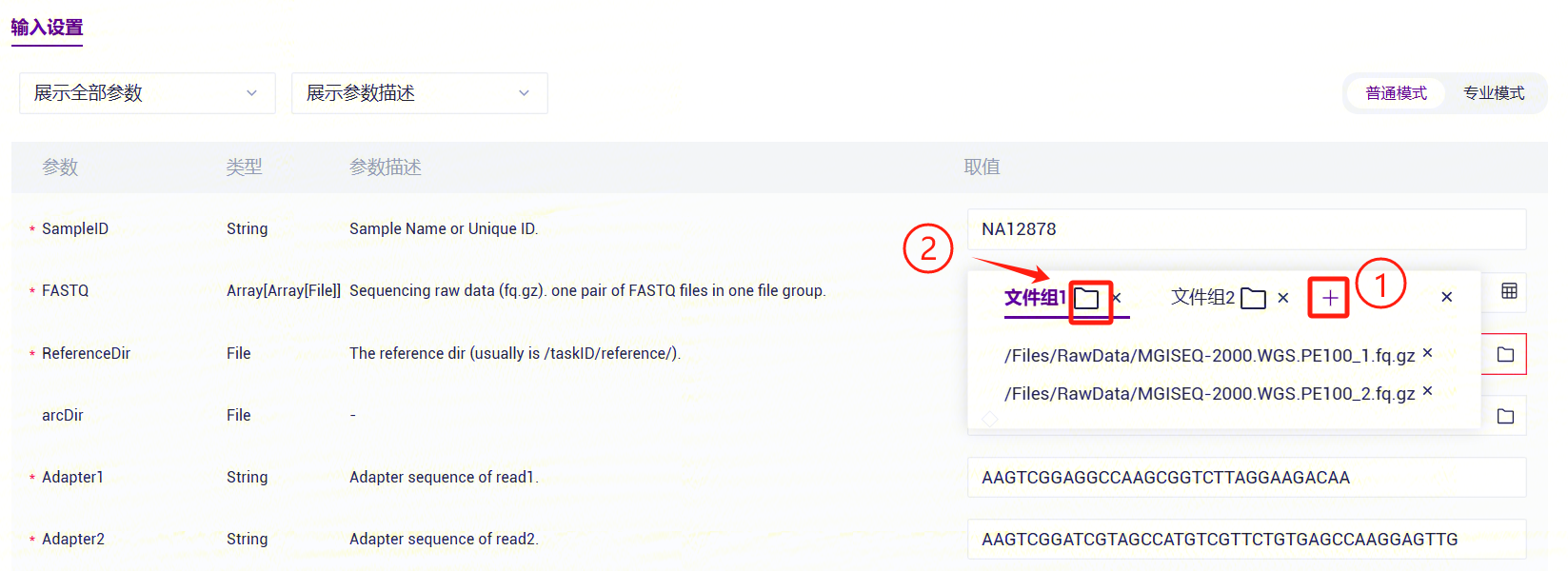
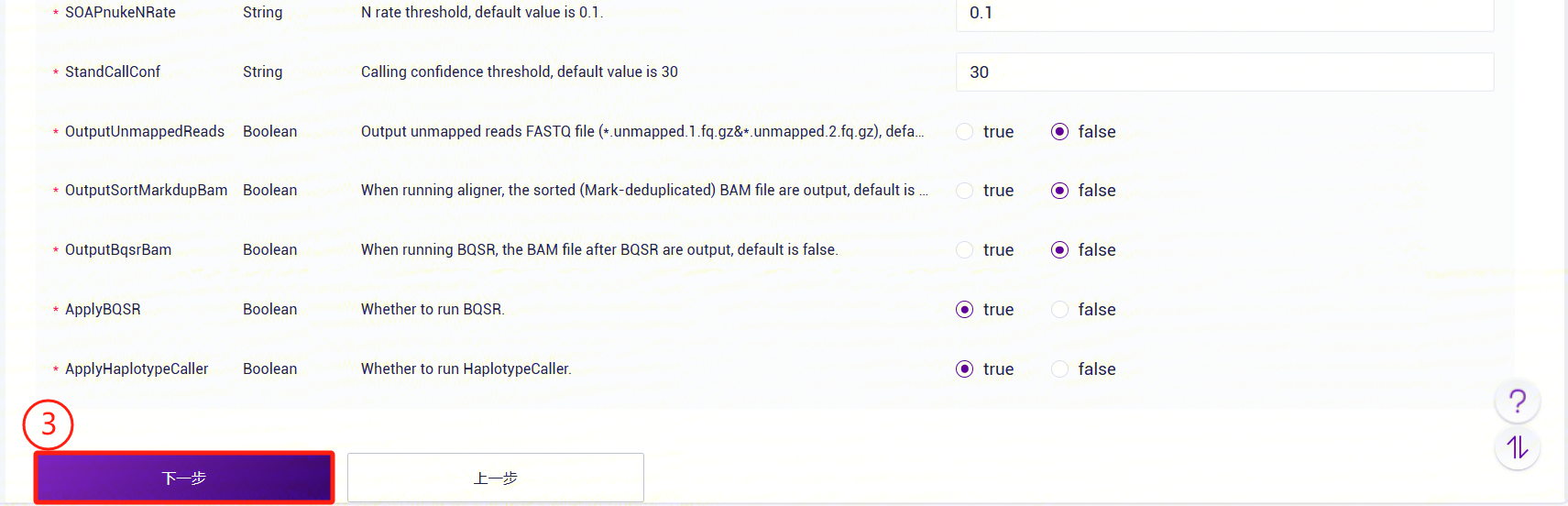
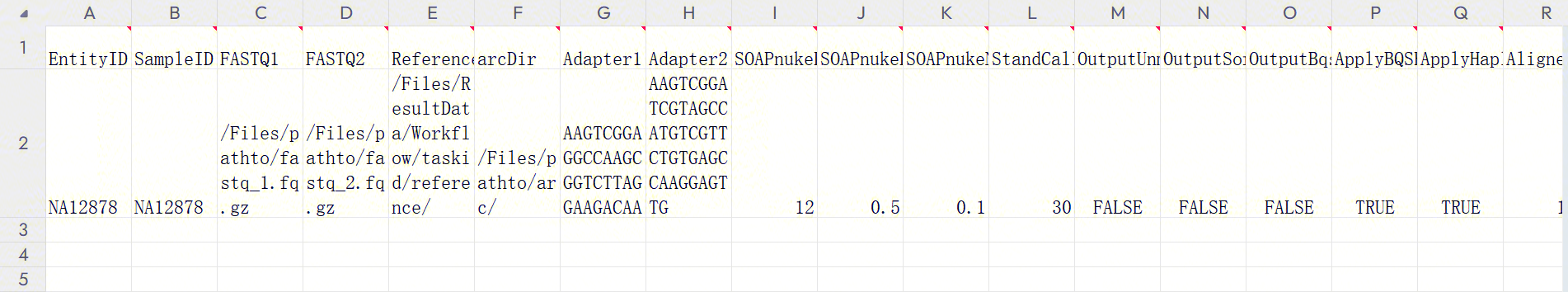
LUSH_Germline_FASTQ_WGS_NHS参数说明:
- SampleID:样本名称或特殊ID,默认与Entity ID一致;
- FASTQ:对原始数据进行测序 (fq.gz或者arc)。PE:一个文件组中有一对 FASTQ 文件。SE:一个文件组中有一个 FASTQ 文件;
- ReferenceDir:支持用户用自己上传的基因组数据构建参考基因组(参考3.2.3),输入为LUSH_reference_index流程结果文件中的 reference 目录;
- arcDir: 用于arcseq解压的FASTQ index文件所在文件夹,如若FASTQ格式为fq.gz可不用填写。
- Adapter1:Read1 的接头序列(用于过滤);
- Adapter2:Read2 的接头序列(用于过滤);
- SOAPnukeLowQual:SOAPnuke过滤的低质量值阈值,默认为12;
- SOAPnukeLowQualityRate:SOAPnuke过滤的低质量值比例阈值,默认为0.5;
- SOAPnukeNRate:SOAPnuke过滤的N占Reads比例阈值,默认为0.1;
- StandCallConf:变异检测置信阈值,默认为30;
- OutputUnmappedReads:是否输出没有比对上的reads(fastq文件),默认不输出;
- OutputSortMarkdupBam:是否输出排序去重后的bam文件,默认不输出;
- OutputBqsrBam:是否输出BQSR后的bam文件,默认不输出;
- ApplyBQSR:是否运行 BQSR,默认情况下运行。如果选择“否”,将使用排序去重后的 BAM 文件进行变异检测。如果参考基因组的 knownsites VCF 为空,将强制设置为“否”。
- ApplyHaplotypeCaller:是否运行HaplotypeCaller,默认情况下运行;
- AlignerMemorySet:比对内存设置,默认情况下128(G);
- BQSRMemorySet:BQSR内存设置,默认情况下64(G);
- HaplotypeCallerMemorySet:HaplotypeCaller内存设置,默认情况下64(G)。
- 点击【运行】启动分析,等待结果输出(图3-15)
3.3 使用场景二:LUSH一口价流程手动投递
3.3.1 步骤一:上传数据
与使用场景一(手动投递)步骤一一致(详见3.2.1步骤一:上传数据)。
3.3.2 步骤二:添加工作流
与使用场景一(手动投递)步骤二一致(详见3.2.2步骤二:添加工作流)。
3.3.3 步骤三: 添加公共库hg38参考基因组文件
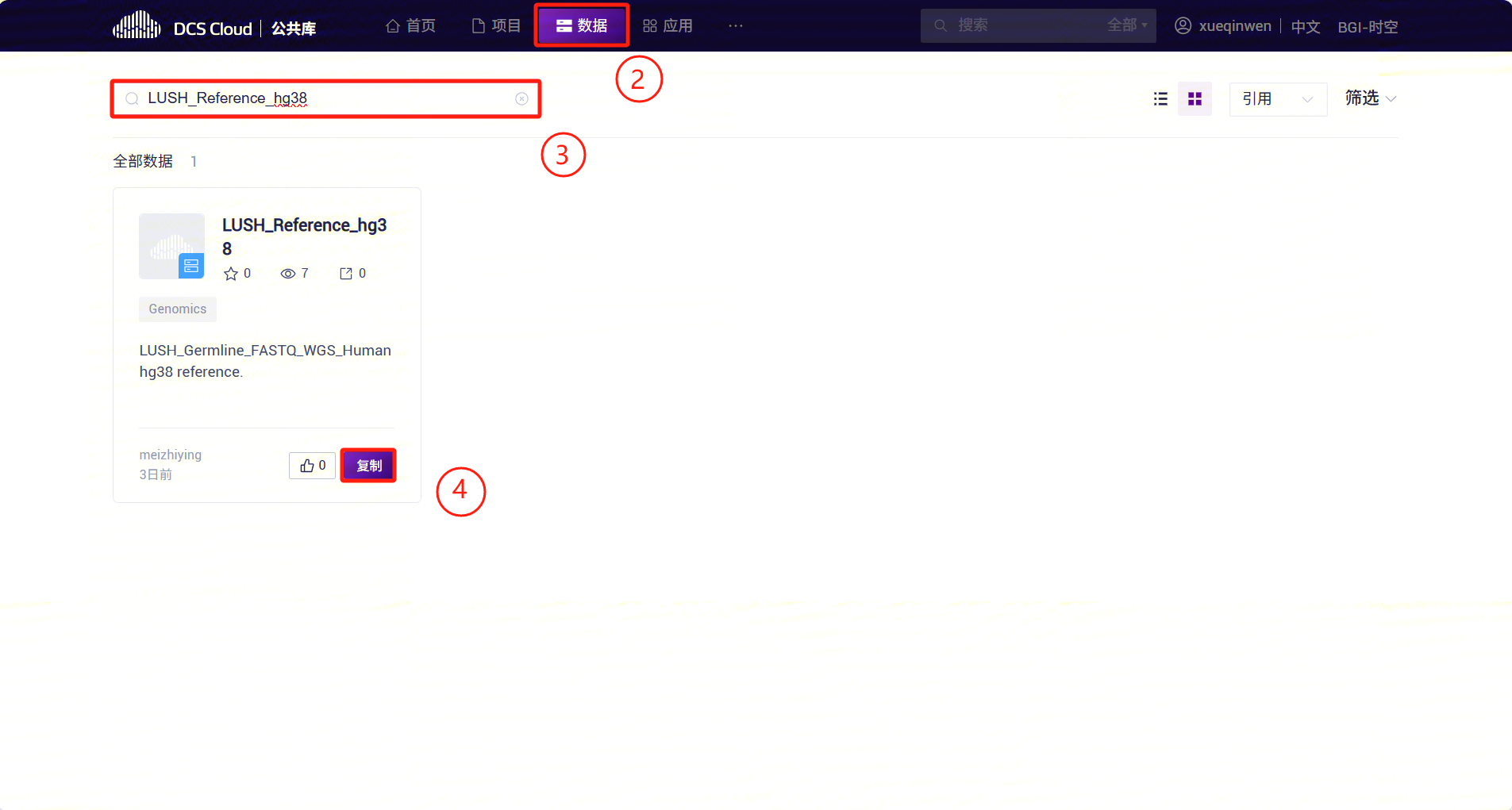
- 点击页面顶部的“公共库”标签,进入公共库界面。

- 在公共库页面中,点击数据标签,进入数据标签页。在搜索栏中输入“LUSH_Reference_hg38”进行检索,并在检索结果中找到LUSH_Reference_hg38卡片,点击复制按钮。
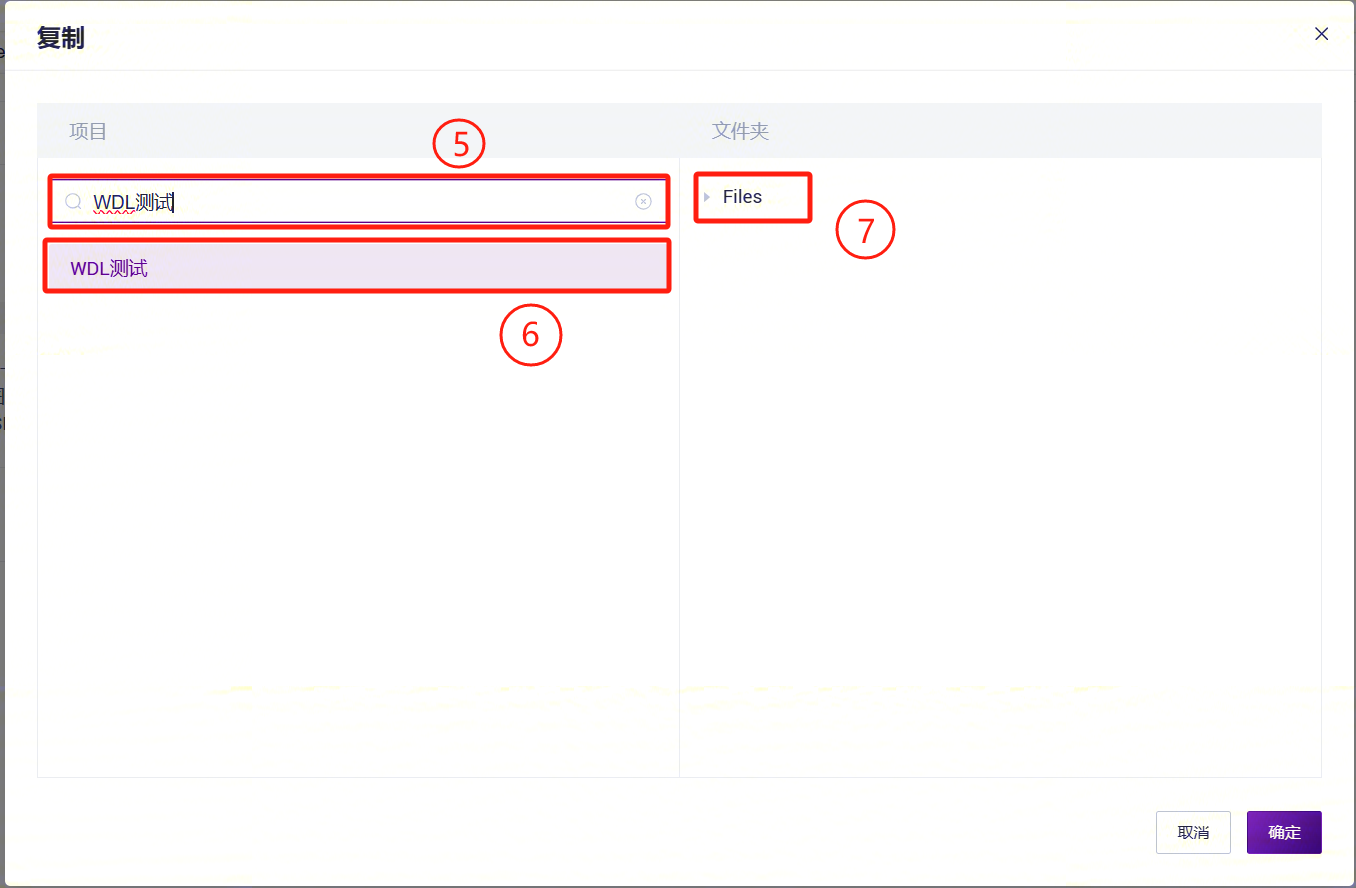
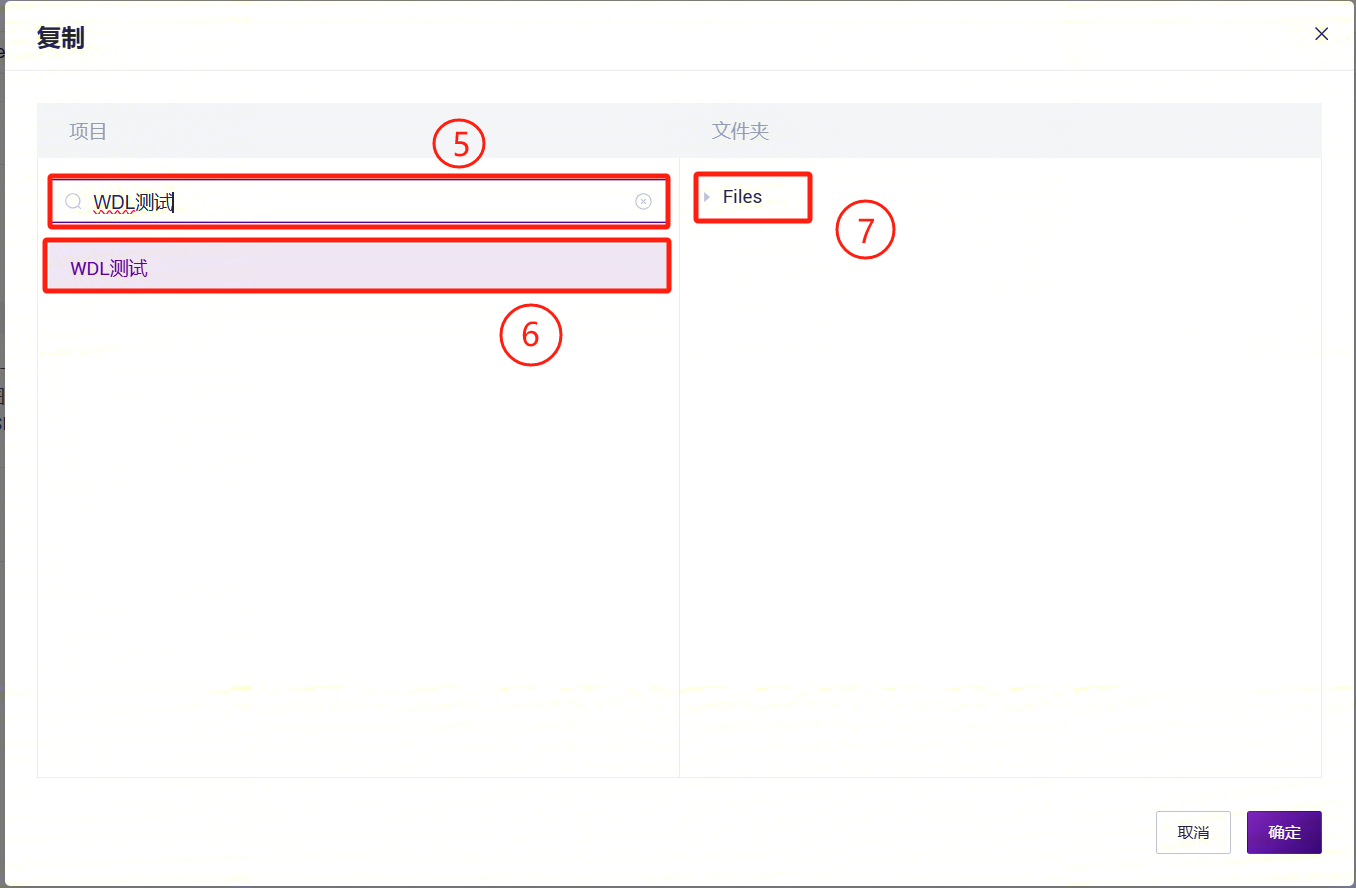
- 在弹出的对话框中,选择您需要存放数据的项目。进入项目下的指定路径,设置数据存放的具体位置,并完成公共数据的拉取。
3.3.4 步骤四:LUSH_Germline_FASTQ_WGS_Human分析流程
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入LUSH_Germline_FASTQ_WGS_Human,点击【运行】(参考3.2.3):
- 输入实体 ID(一般为样品ID),点击【下一步】(参考3.2.3):
- LUSH_Germline_FASTQ_WGS_Human录入样本信息。PE FASTQ 输入:根据成对的 FASTQ 文件数量,点击“+”按钮添加文件组,并在每个文件组中选择对应的 FASTQ1 和 FASTQ2 文件; SE FASTQ输入:操作与 PE 数据类似,每个文件组中放置一个 FASTQ 文件。
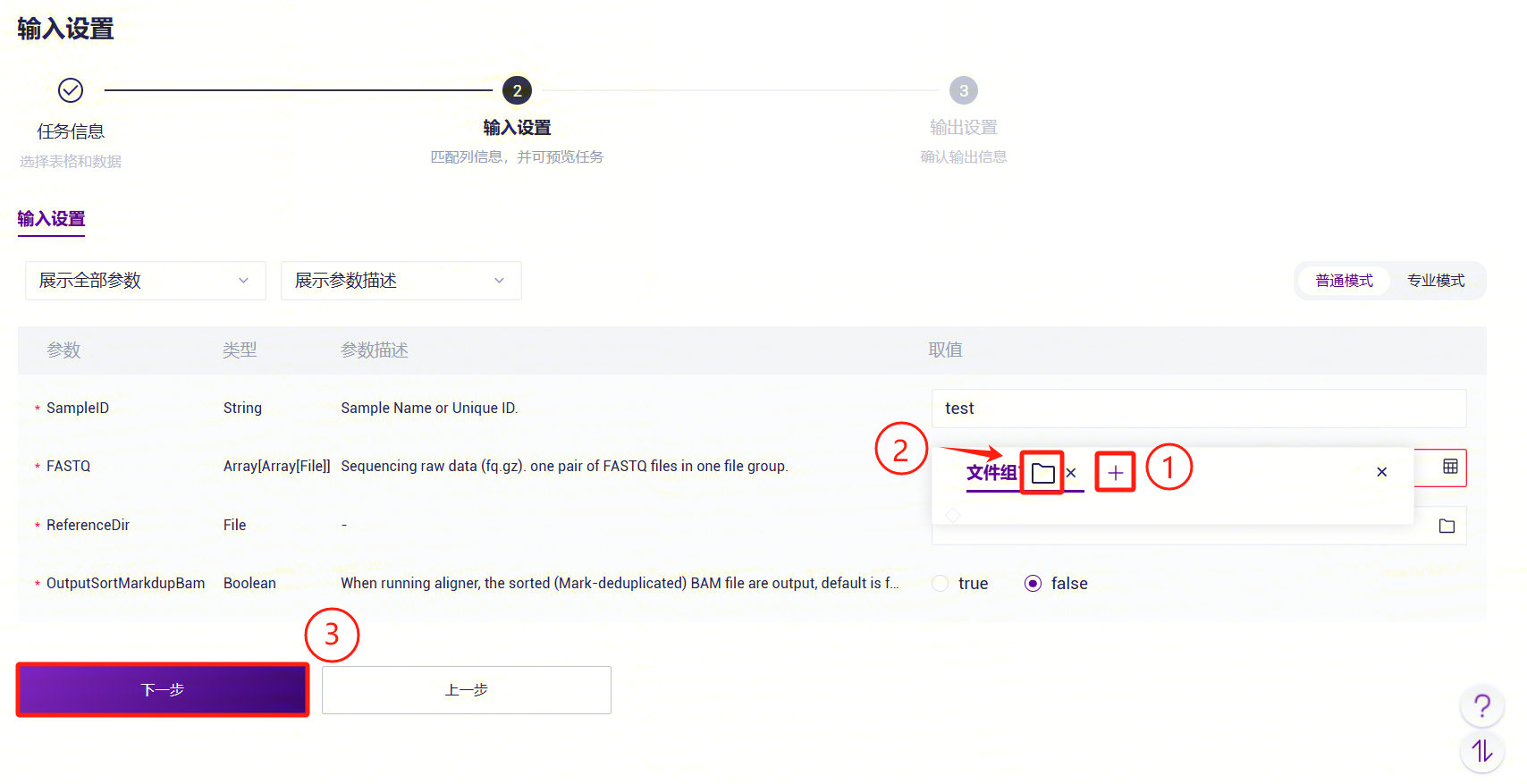
LUSH_Germline_FASTQ_WGS_Human参数说明:
- SampleID:样本名称或特殊ID,默认与Entity ID一致;
- FASTQ:对原始数据进行测序 (fq.gz或者arc)。PE:一个文件组中有一对 FASTQ 文件。SE:一个文件组中有一个 FASTQ 文件;
- ReferenceDir:参考基因组文件夹,需要从公共库中拉取至分析项目中;
- OutputSortMarkdupBam:是否输出排序去重后的bam文件,默认不输出;
- 点击【运行】启动分析,等待结果输出(图3-20)
3.4 使用场景三:ExpansionHunter_WGS_STR流程手动投递
ExpansionHunter_WGS_STR流程使用Expansion Hunter进行STR序列的查找。Expansion Hunter 是一款用于对短串联重复序列和侧翼变异进行靶向基因分型的工具。它通过对 BAM/CRAM 文件进行靶向搜索来查找跨越、侧翼和完全包含在每个重复序列中的reads。该分析流程当前仅针对基于人类基因组hg38参考序列构建的BAM/CRAM格式比对数据提供支持。
3.4.1 步骤一:输入数据
需要在LUSH_Germline_FASTQ_WGS_Human 分析时,将OutputSortMarkdupBam 参数设置为true,使流程结果文件输出aligner bam。
3.4.2 步骤二:添加工作流
与使用场景一(手动投递)步骤二类似,根据流程名进行检索(详见3.2.2步骤二:添加工作流)。
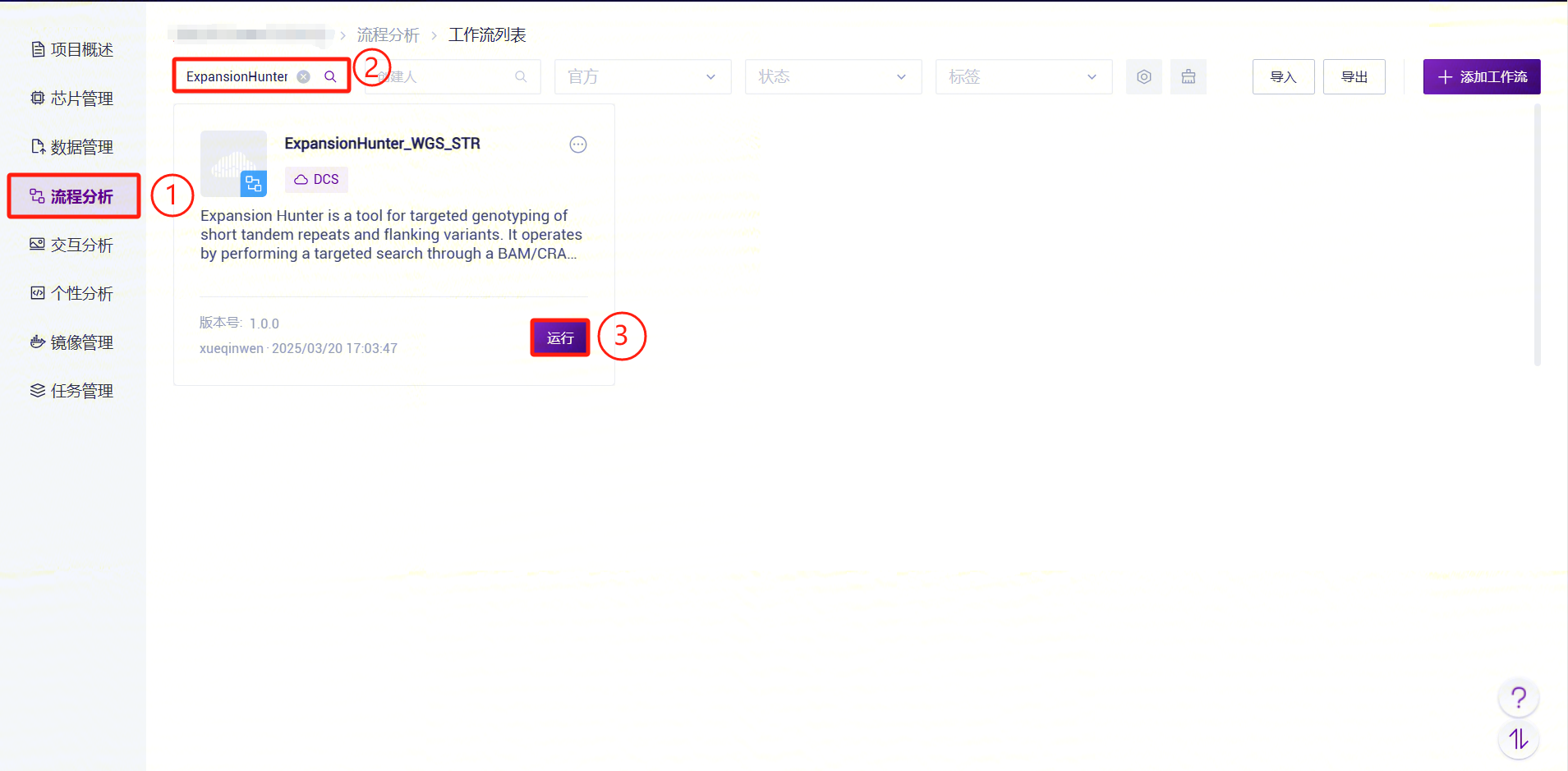
3.4.3 步骤三:ExpansionHunter_WGS_STR分析流程
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入ExpansionHunter_WGS_STR,点击【运行】(参考3.2.3):
- 输入实体 ID(一般为样品ID),点击【下一步】(参考3.2.3):
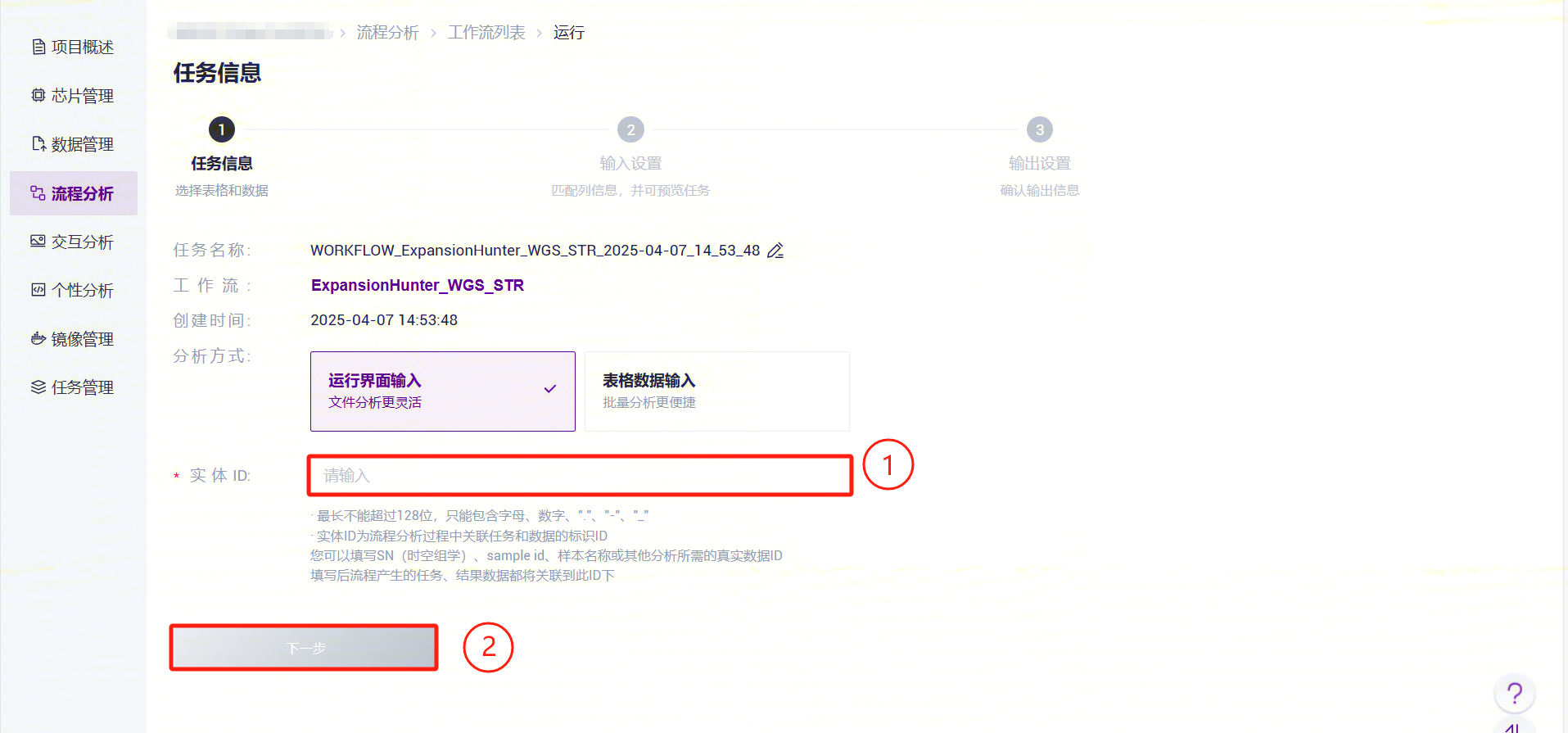
- ExpansionHunter_WGS_STR录入样本信息(图3-23)。
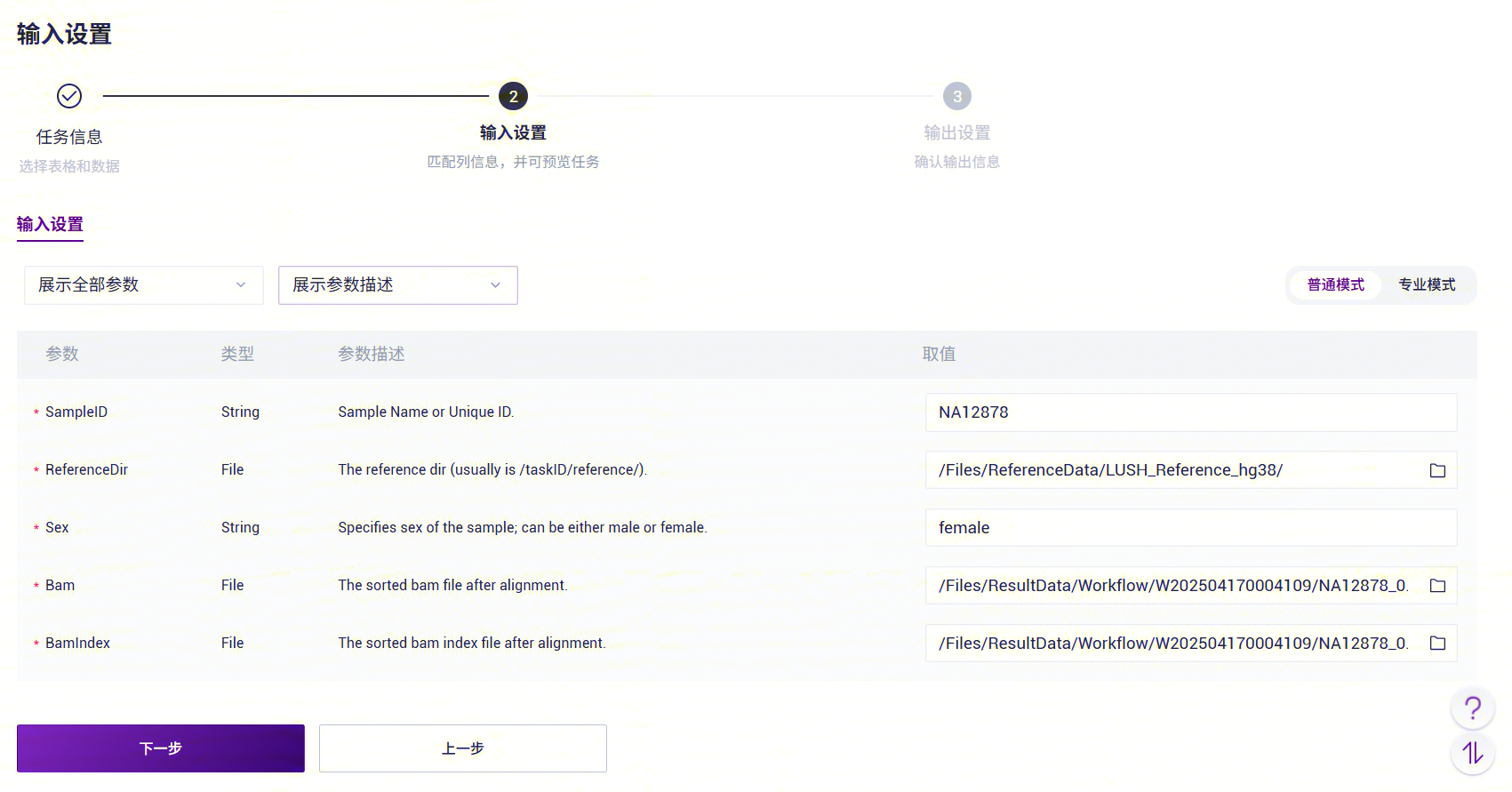
ExpansionHunter_WGS_STR参数说明:
- SampleID:样本名称或特殊ID,默认与Entity ID一致;
- ReferenceDir:参考基因组文件夹,需要从公共库中拉取至分析项目中;
- Sex: 样本性别,male 或者female。
- Bam:比对去重排序后的bam文件;
- BamIndex: 比对去重排序后的bam文件的索引文件。
- 点击【运行】启动分析,等待结果输出
3.5 使用场景四:PanGenie_WGS_SV流程手动投递
PanGenie_WGS_SV流程是基于图谱的泛基因组工作流程,专为使用PanGenie从测序数据进行基因型插补和结构变异(SV)检测而设计。基因型的计算基于读取的k-mer计数和一组已知的、完全组装的单倍型。与基于比对的方法相比,PanGenie在几乎所有测试的变异类型和覆盖度下均实现了更高的基因型一致性。对于大片段插入(≥50 bp)和重复区域的变异,改进尤为显著,使得这些类别的变异能够被纳入全基因组关联研究中。
3.5.1 步骤一:上传数据
与使用场景一(手动投递)步骤一一致(详见3.2.1步骤一:上传数据)。
3.5.2 步骤二:添加工作流
与使用场景一(手动投递)步骤二类似,根据流程名进行检索(详见3.2.2步骤二:添加工作流)。
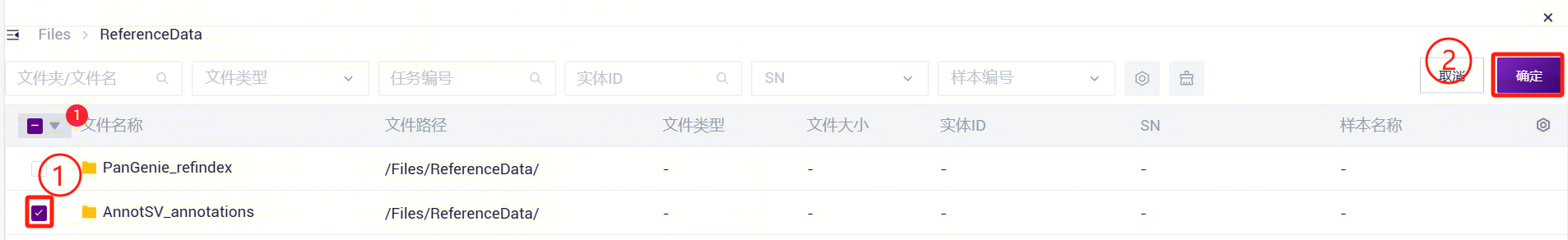
3.5.3 步骤三:添加PanGenie参考基因组文件
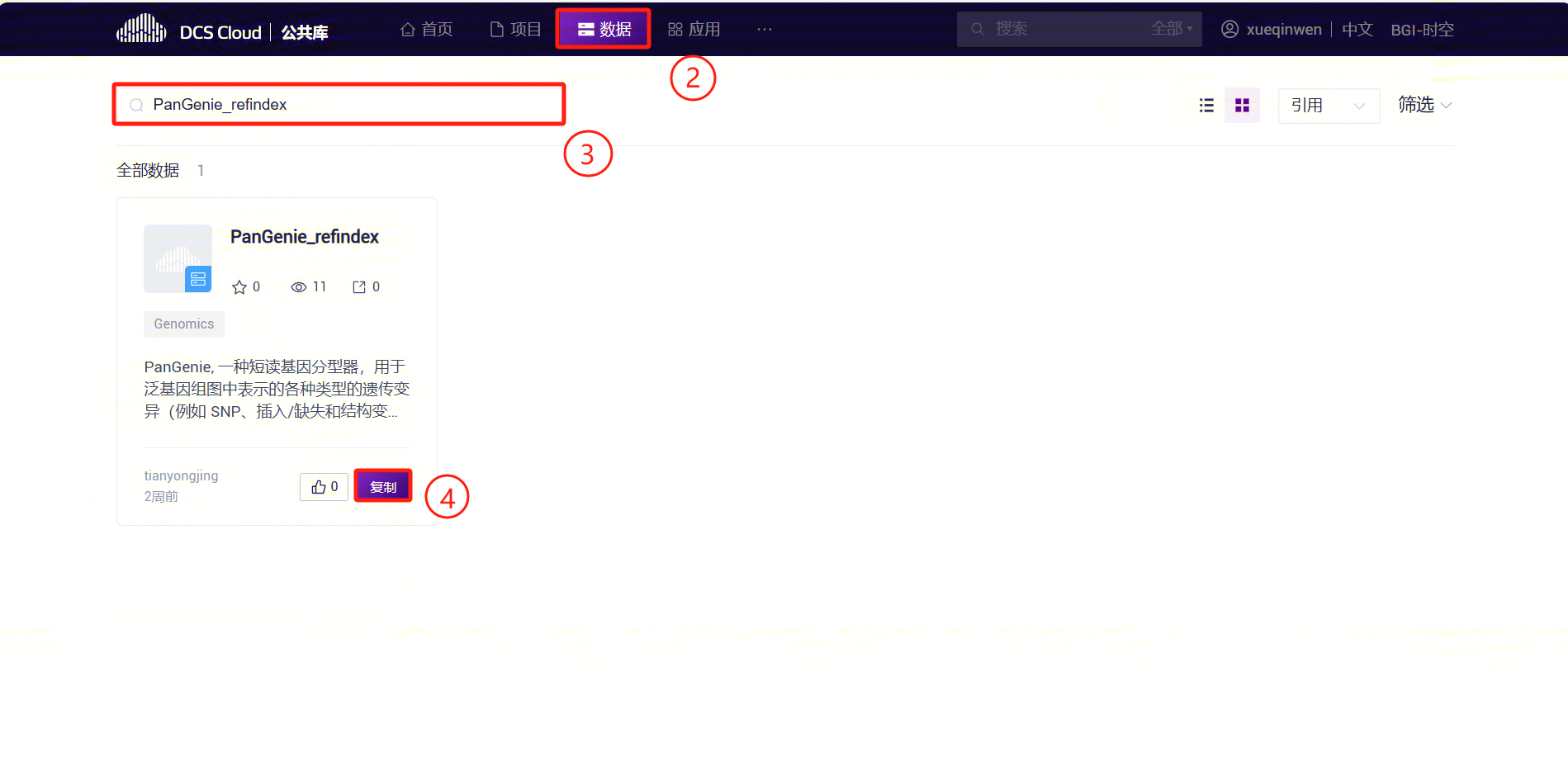
- 点击页面顶部的“公共库”标签,进入公共库界面。

- 在公共库页面中,点击数据标签,进入数据标签页。在搜索栏中输入“PanGenie_refindex”进行检索,并在检索结果中找到PanGenie_refindex卡片,点击复制按钮。
- 在弹出的对话框中,选择您需要存放数据的项目。进入项目下的指定路径,设置数据存放的具体位置,并完成公共数据的拉取。
3.5.4 步骤四:添加人类结构变异注释数据库文件
按照步骤三的操作方法,将检索目标更改为AnnotSV_annotations。
3.5.5 步骤五:PanGenie_WGS_SV分析流程
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入PanGenie_WGS_SV,点击【运行】(参考3.2.3):
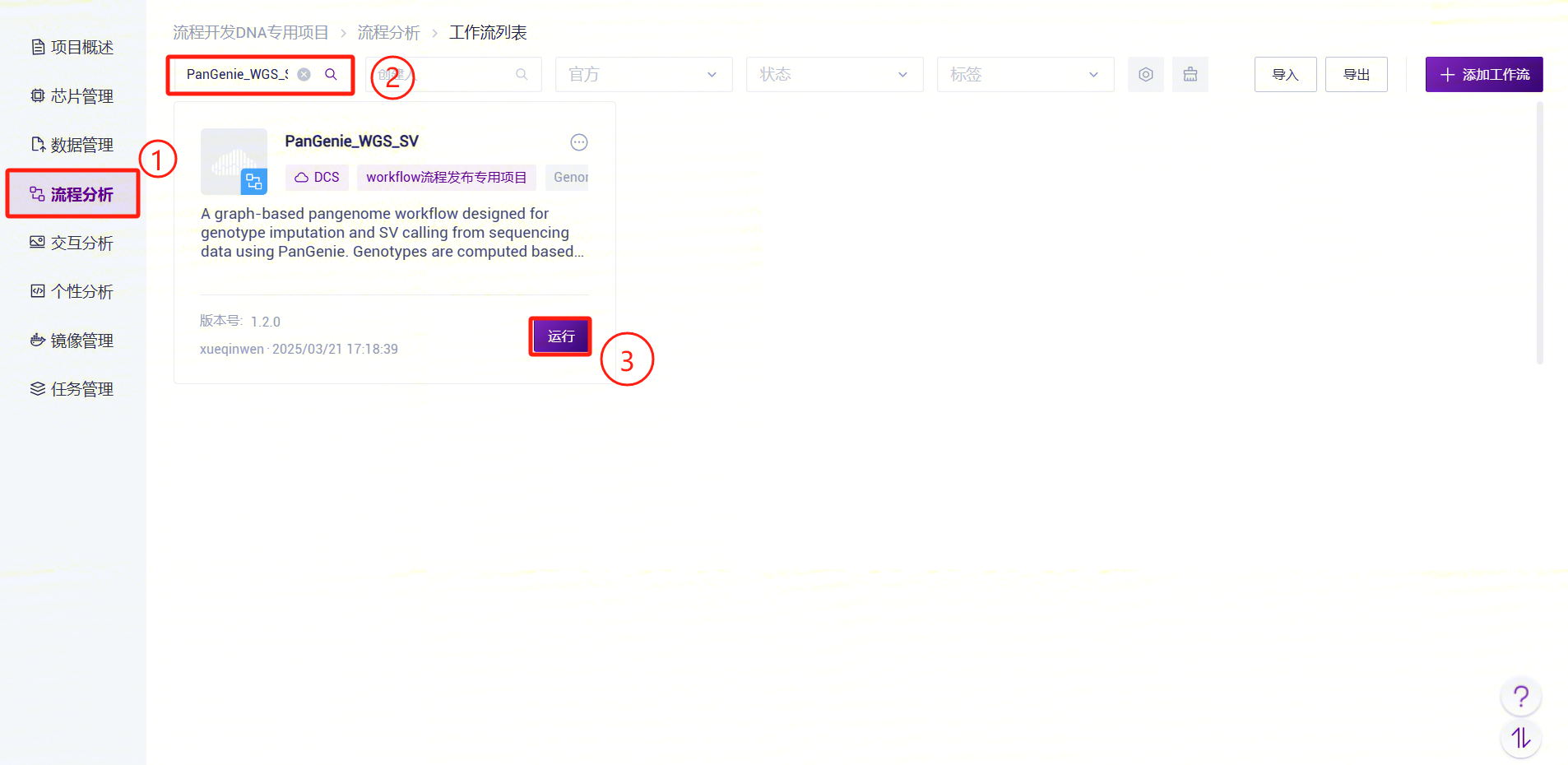
- 输入实体 ID(一般为样品ID),点击【下一步】(参考3.2.3):
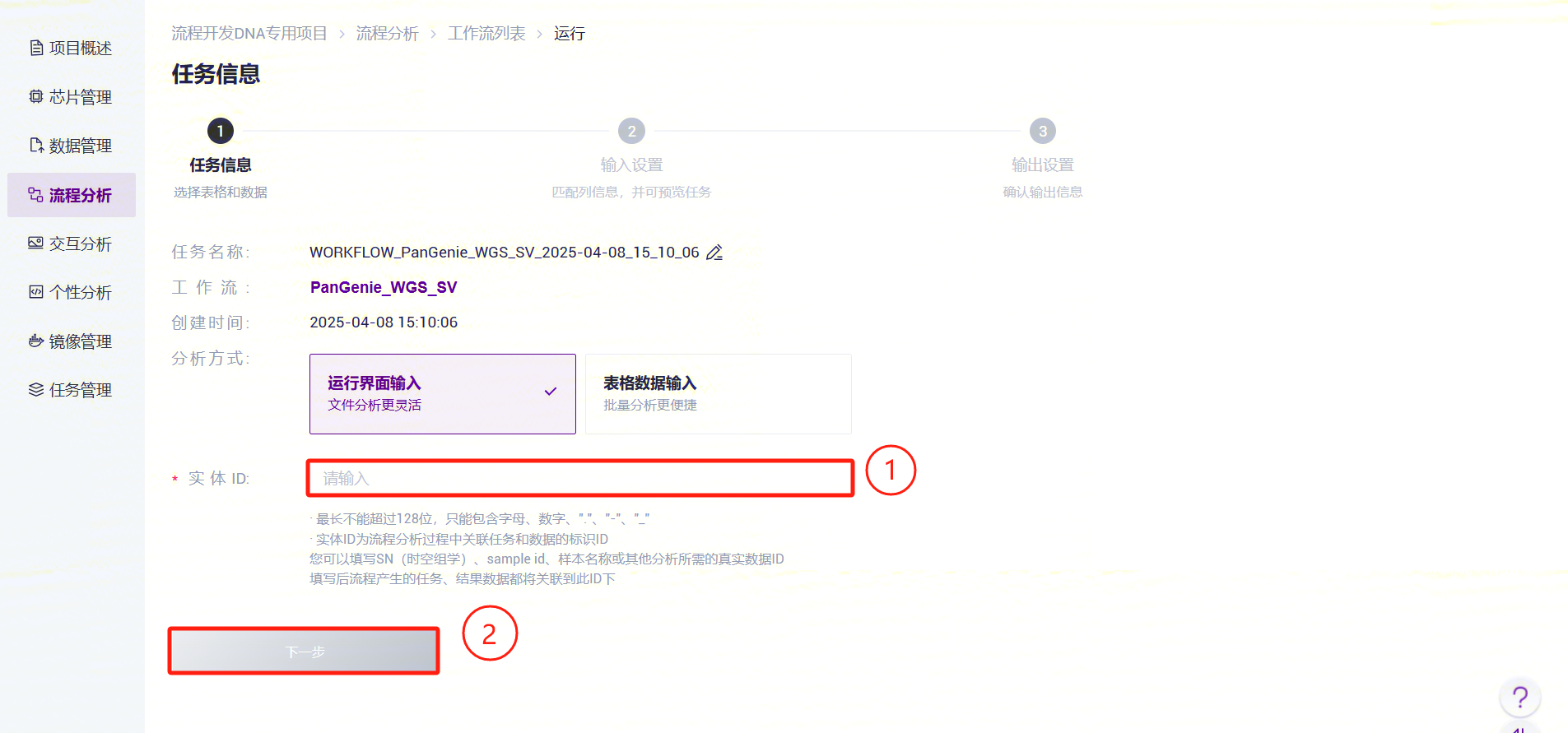
- PanGenie_WGS_SV录入样本信息。在FqFile点击文件夹图案,进入数据管理页面,选择FASTQ文件。
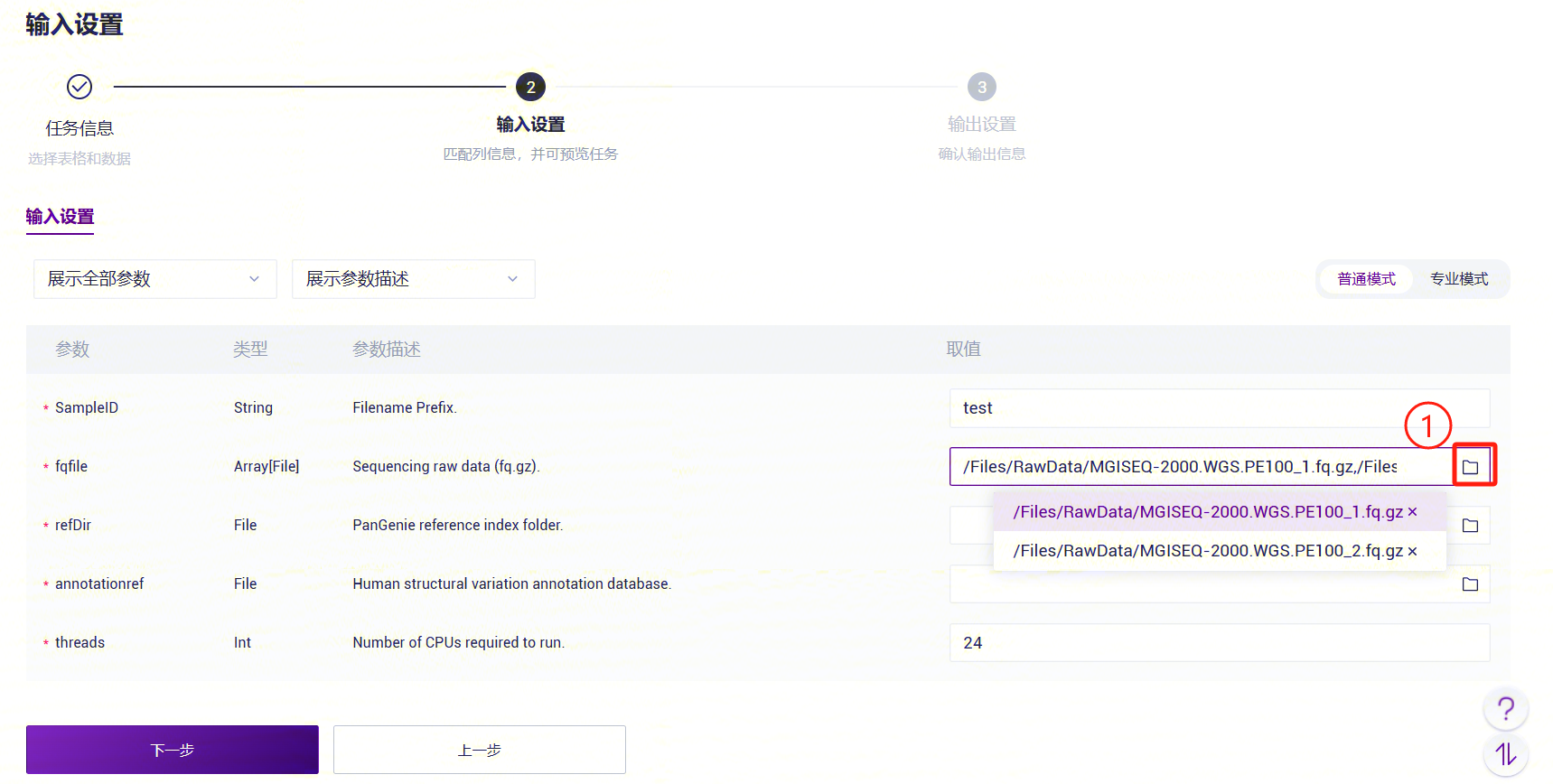
- PanGenie_WGS_SV录入PanGenie参考基因组文件夹。在RefDir参数点击文件夹图案,进入数据管理页面,选择PanGenie参考基因组文件夹。
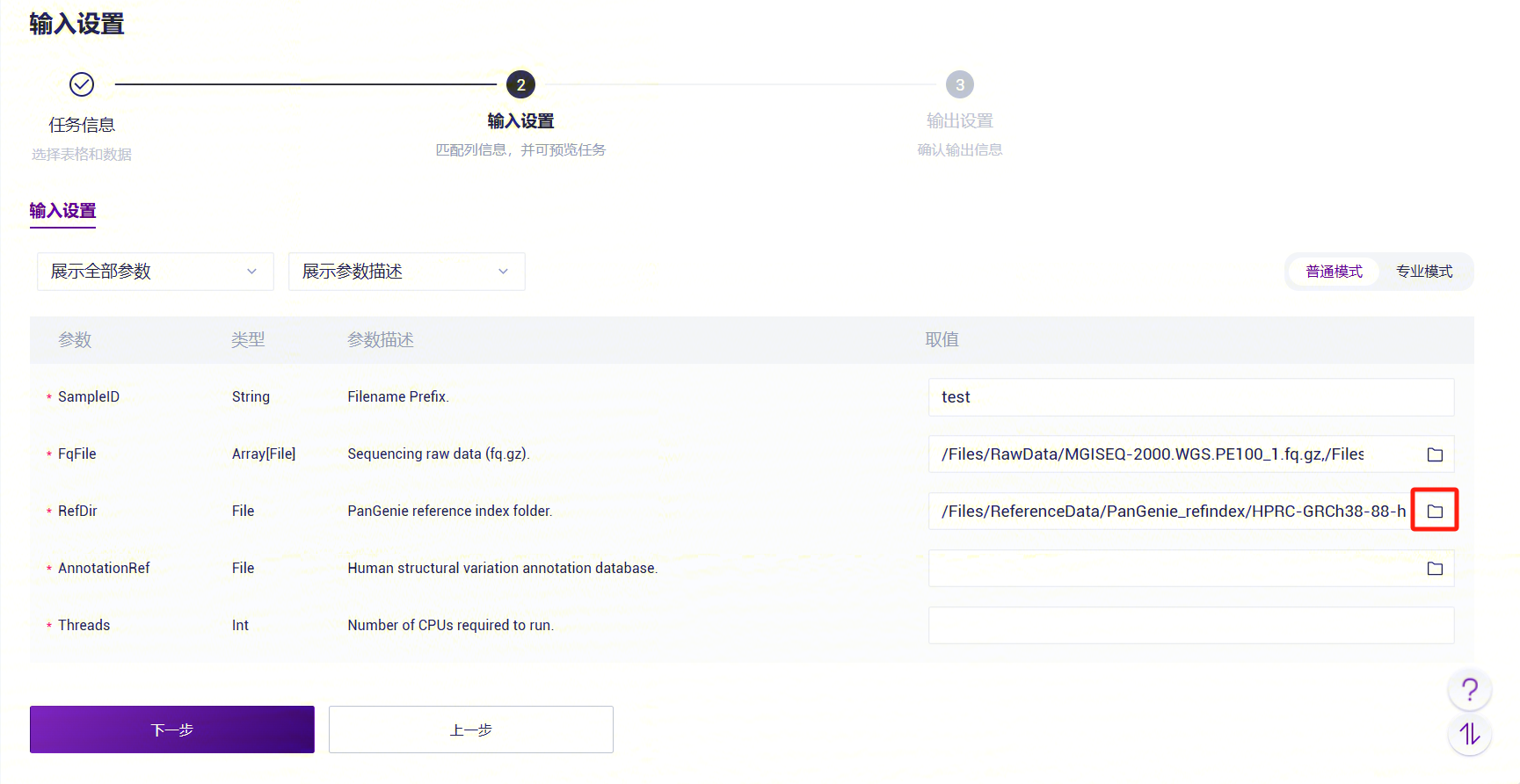
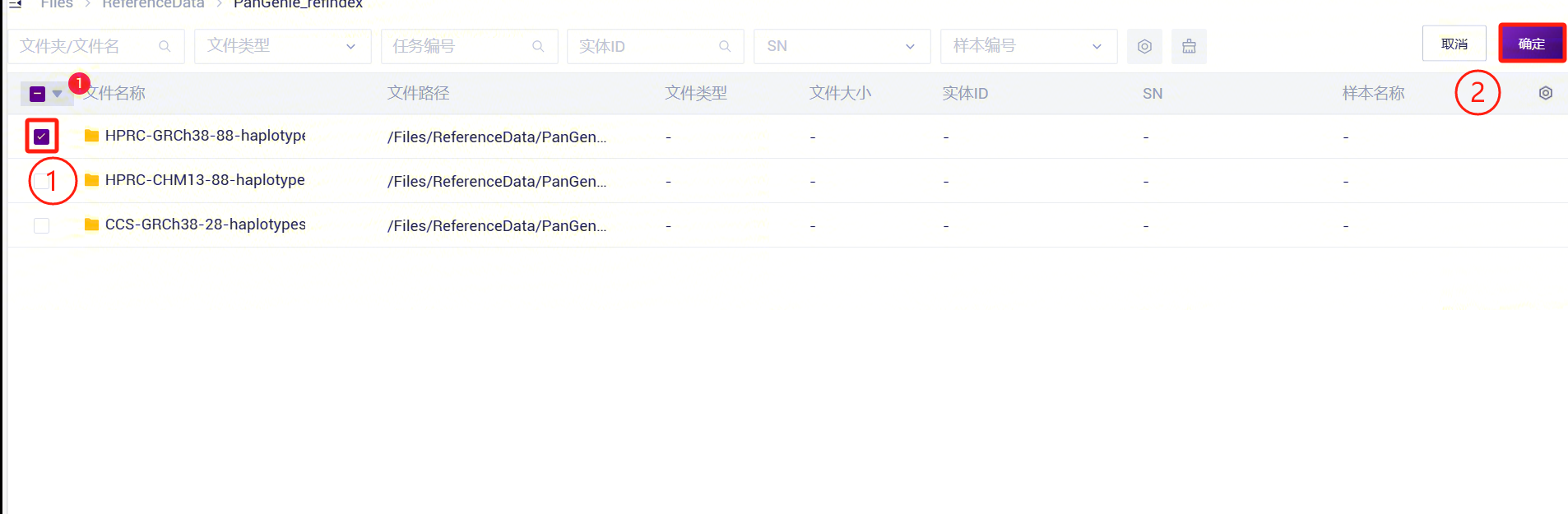
- PanGenie_WGS_SV录入人类结构变异注释数据库文件夹。在AnnotationRef参数点击文件夹图案,进入数据管理页面,选择人类结构变异注释数据库。
PanGenie_WGS_SV参数说明:
- SampleID:样本名称或特殊ID,默认与Entity ID一致;
- FqFile:测序文件;
- RefDir: PanGenie参考基因组文件;
- AnnotationRef:人类结构变异注释数据库;
- Threads:任务运行CPU核数设置。
- 点击【运行】启动分析,等待结果输出(图3-33)
3.6 使用场景五:CNVpytor_WGS_CNV流程手动投递
CNVpytor_WGS_CNV流程是基于CNVpytor的CNV检测与分析流程,用于从测序数据中检测和分析拷贝数变异(CNV)。CNVpytor继承了其前身重新实现的核心引擎,并扩展了可视化、模块化、性能和功能。此外,CNVpytor利用来自单核苷酸多态性(SNP)和小片段插入/缺失(indel)数据的B等位基因频率(B-allele frequency)似然信息,作为CNV/拷贝数变异(CNA)的额外证据,并作为拷贝数中性杂合性缺失(copy number-neutral loss of heterozygosity)的主要信息。
3.6.1 步骤一:输入数据
需要在LUSH_Germline_FASTQ_WGS_Human 分析时,将OutputSortMarkdupBam 参数设置为true,使流程结果文件输出aligner bam。
3.6.2 步骤二:添加工作流
与使用场景一(手动投递)步骤二类似,根据流程名进行检索(详见3.2.2步骤二:添加工作流)。
3.6.3 步骤三:CNVpytor_WGS_CNV分析流程
- 点击左侧导航栏【流程分析】进入流程分析页面,在搜索框输入CNVpytor_WGS_CNV,点击【运行】(参考3.2.3):
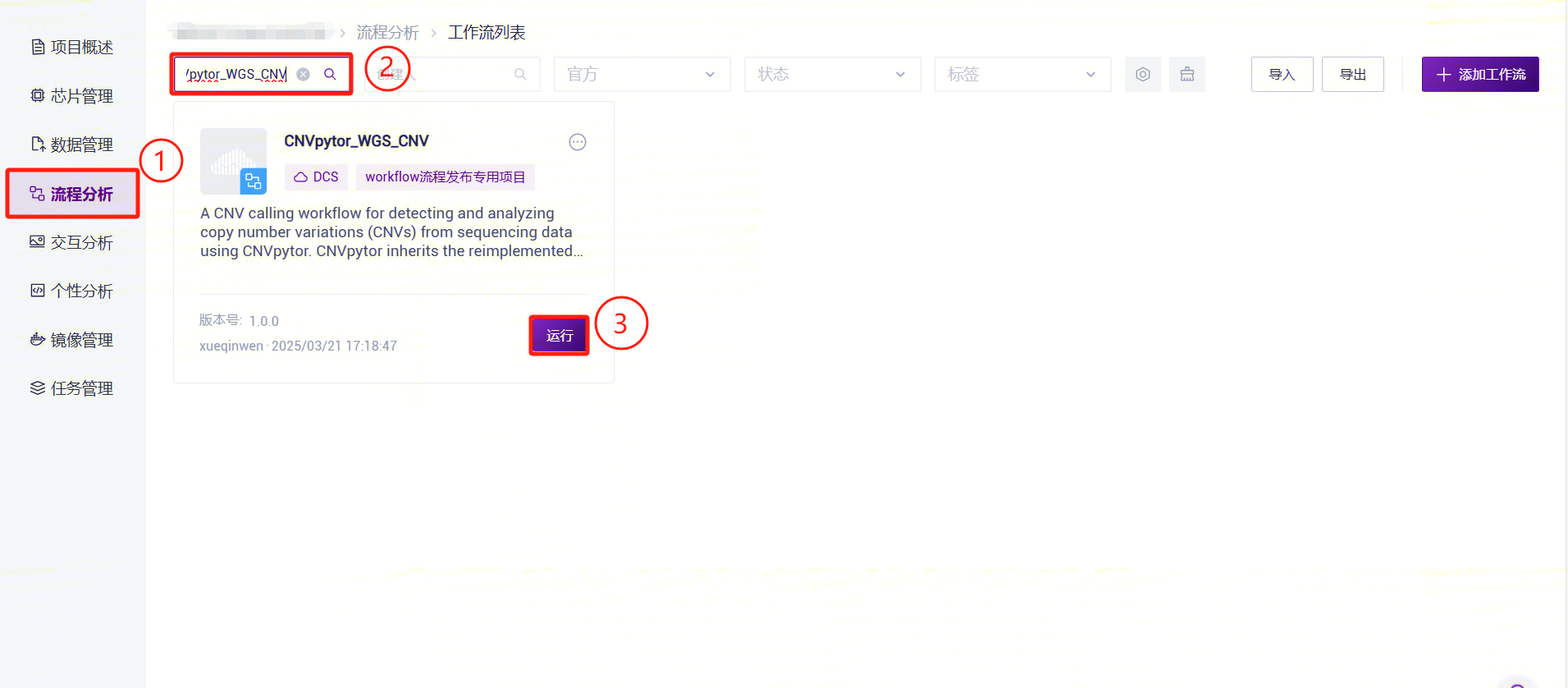
- 输入实体 ID(一般为样品ID),点击【下一步】(参考3.2.3):
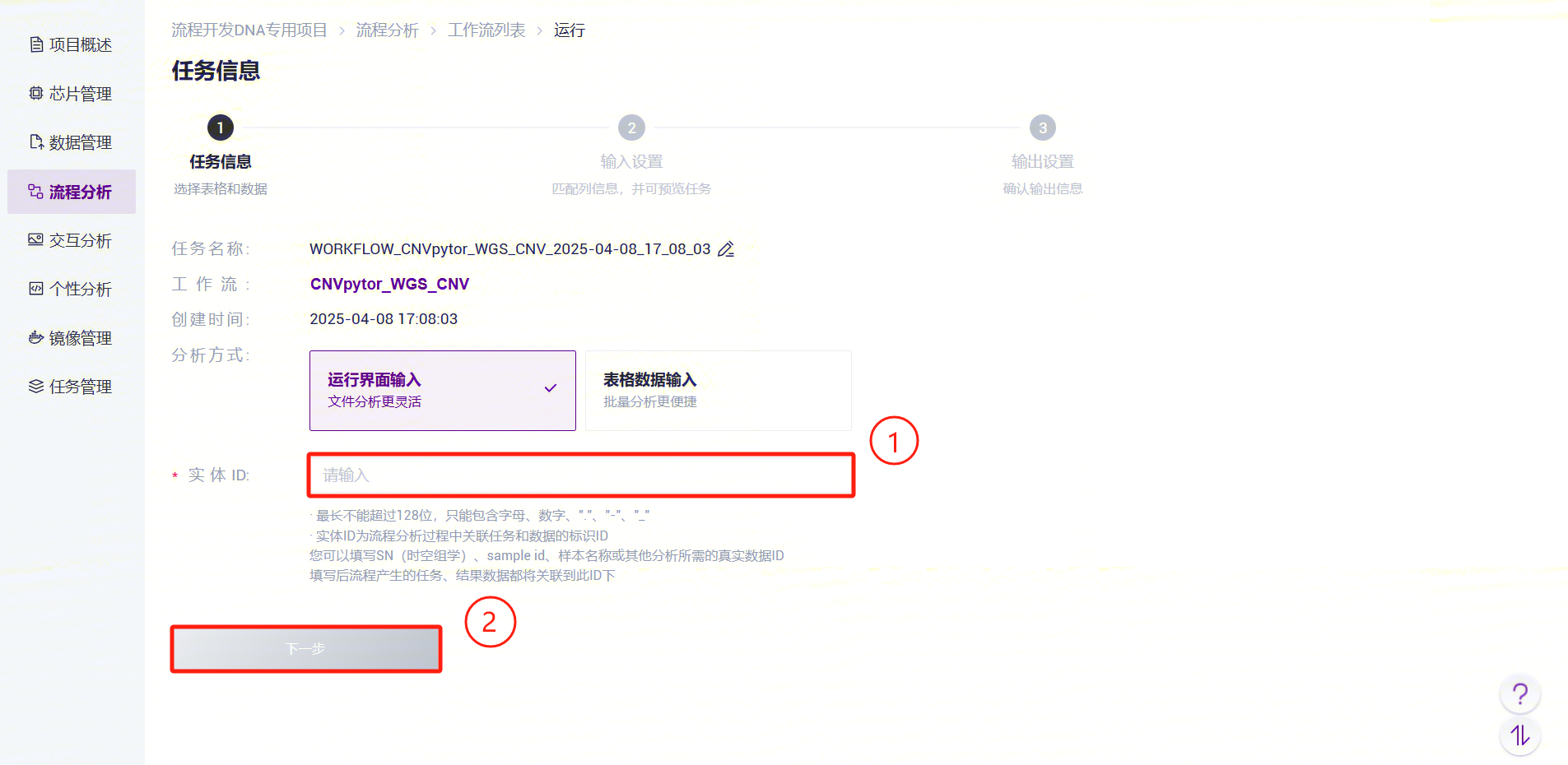
- 在Bam点击文件夹图案,进入数据管理页面,选择Bam文件。
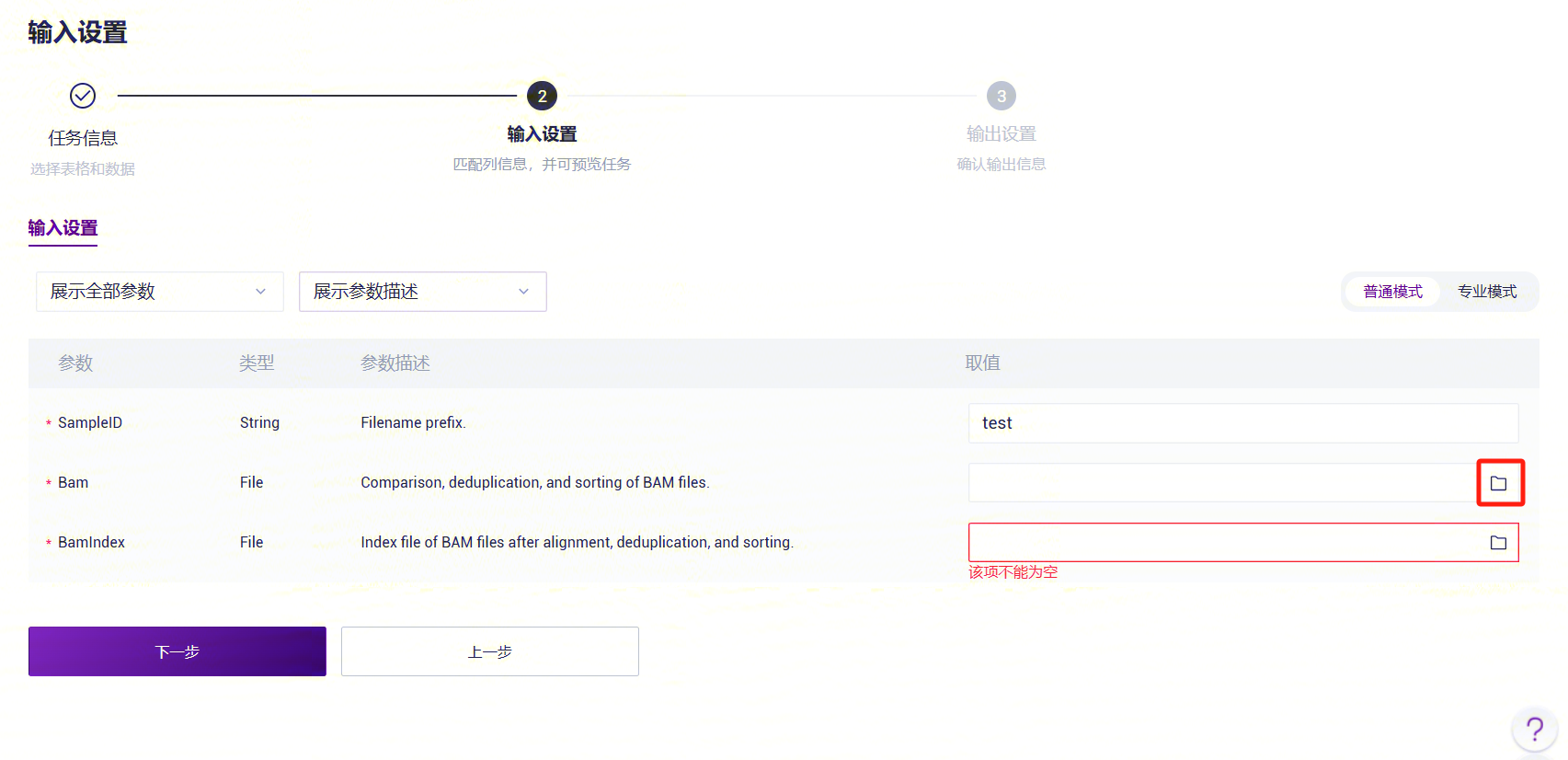
- 在BamIndex点击文件夹图案,进入数据管理页面,选择Bam的索引文件。
CNVpytor_WGS_CNV参数说明:
- SampleID:样本名称或特殊ID,默认与Entity ID一致;
- Bam:比对去重排序后的bam文件;
- BamIndex: 比对去重排序后的bam文件的索引文件;
- 点击【运行】启动分析,等待结果输出(图3-37)
3.7 使用场景六:表格投递(以LUSH_Germline_FASTQ_WGS_NHS为例)
操作共包括五个步骤:上传数据、添加工作流、下载表格模板、填写并导入表格、启动分析。完成样本模板导入后,可批量运行任务,当客户在看到任务状态为![]() 完成时,代表任务已完成,即可查看结果部分(详见3.4部分)。
完成时,代表任务已完成,即可查看结果部分(详见3.4部分)。
3.7.1 步骤一:上传数据
与使用场景一(手动投递)步骤一一致(详见3.2.1步骤一:上传数据)。
3.7.2 步骤二:添加工作流
与使用场景一(手动投递)步骤二一致(详见3.2.2步骤二:添加工作流)。
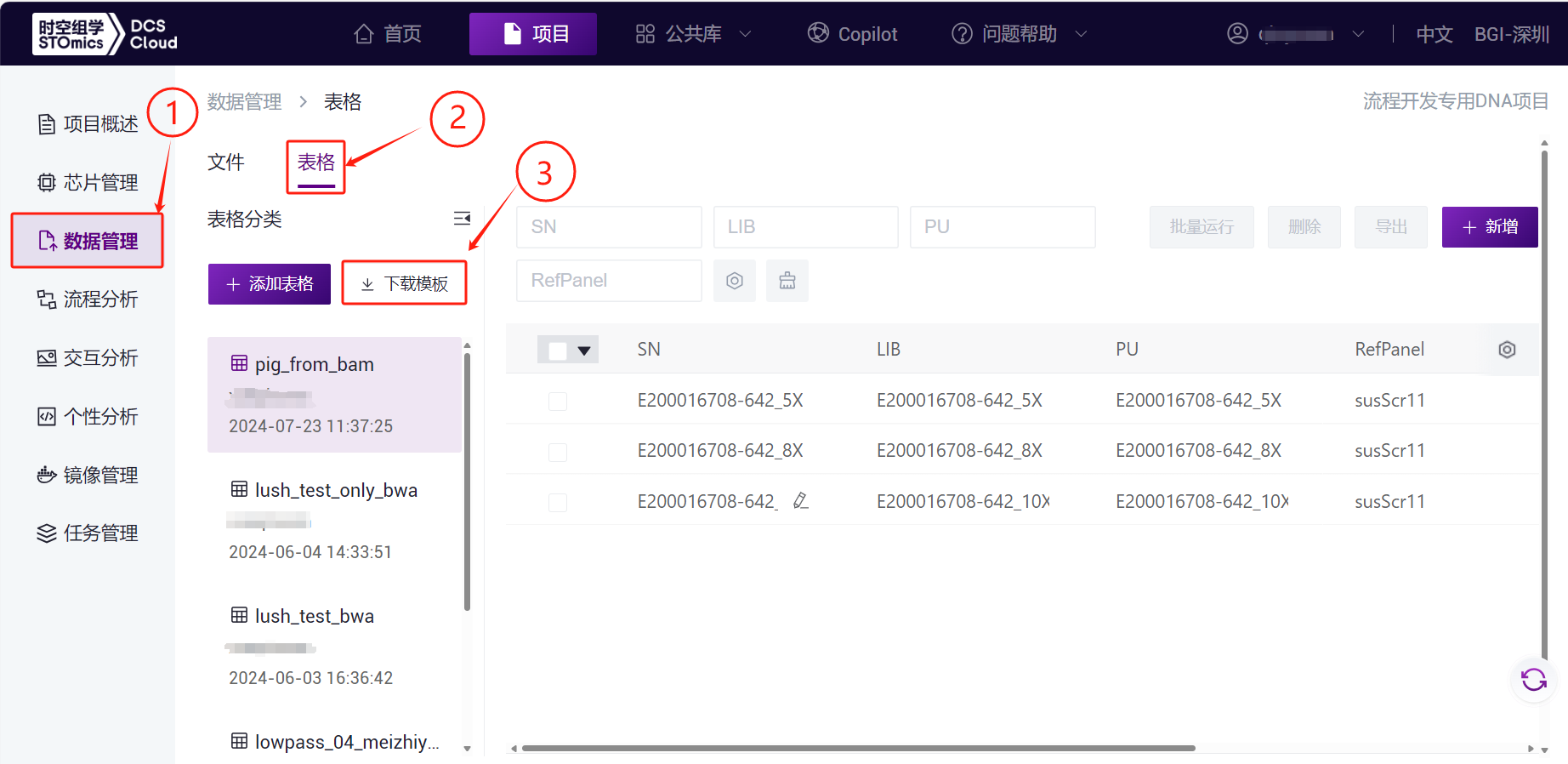
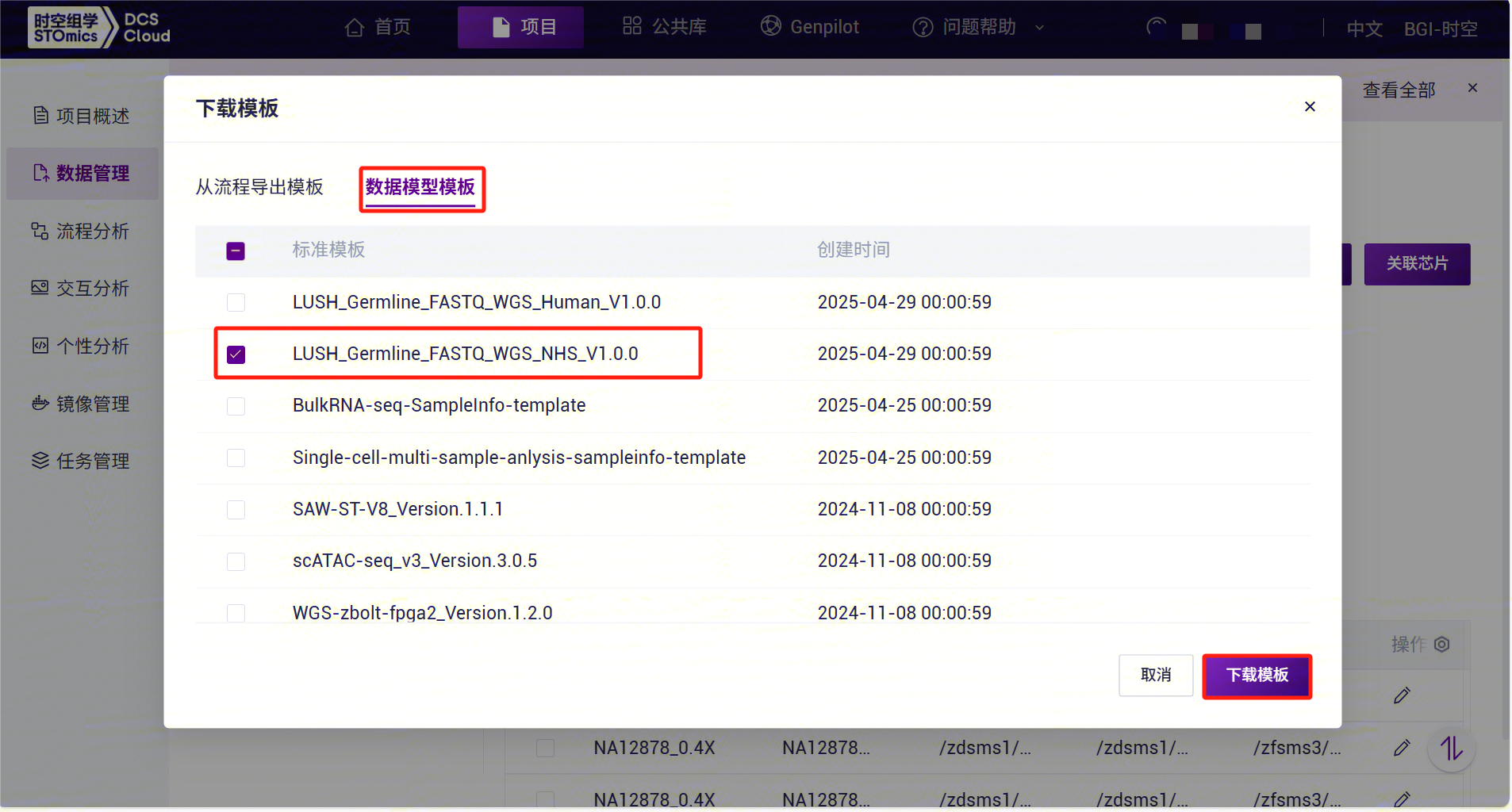
3.7.3 步骤三:下载表格模板
- 点击左侧导航栏【数据管理】,选择【表格】-【下载模板】(如图3-38), 点击【数据模板模型】,选择LUSH_Germline_FASTQ_WGS_NHS_V1.0.0模板下载(LUSH_Germline_FASTQ_WGS_Human流程模板为LUSH_Germline_FASTQ_WGS_Human_V1.0.0):
- 打开后样本模板Excel如图3-39:
3.7.4 步骤四:填写并导入表格
- 该使用场景条件下,导入表格需填写工作表。该场景表示对已测序完成的样本数据在导入表格后,直接进入分析。
Excel注意事项:
[1] 导入文件路径必须在云平台已存在。
[2] Excel中不能合并单元格,单元格内容前后不能有空格或特殊字符。
[3] 分析样本录入(图3-40):
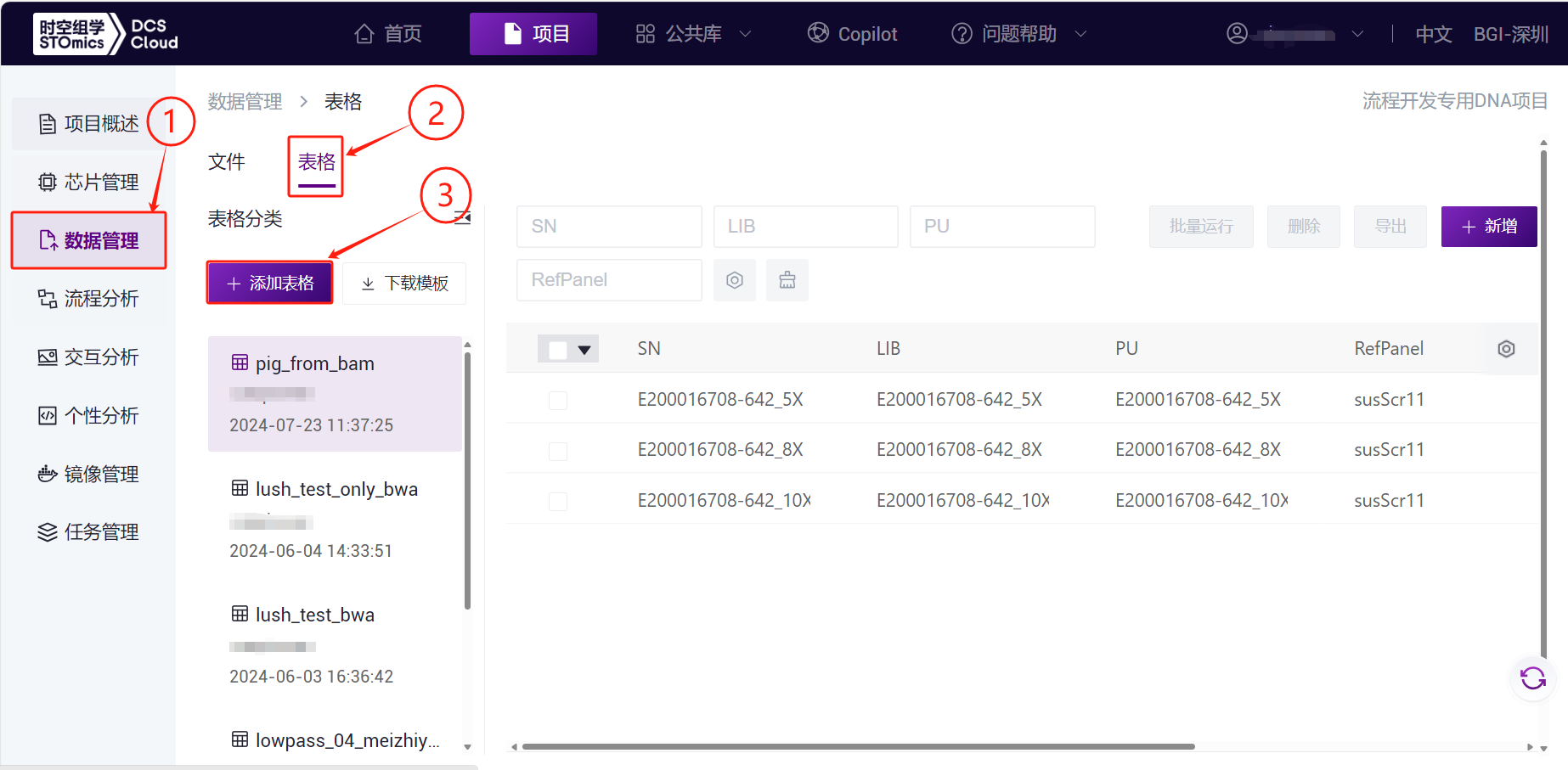
- 配置好表格模板后,回到【数据管理】界面,点击【表格】-【+ 添加表格】(图3-41):
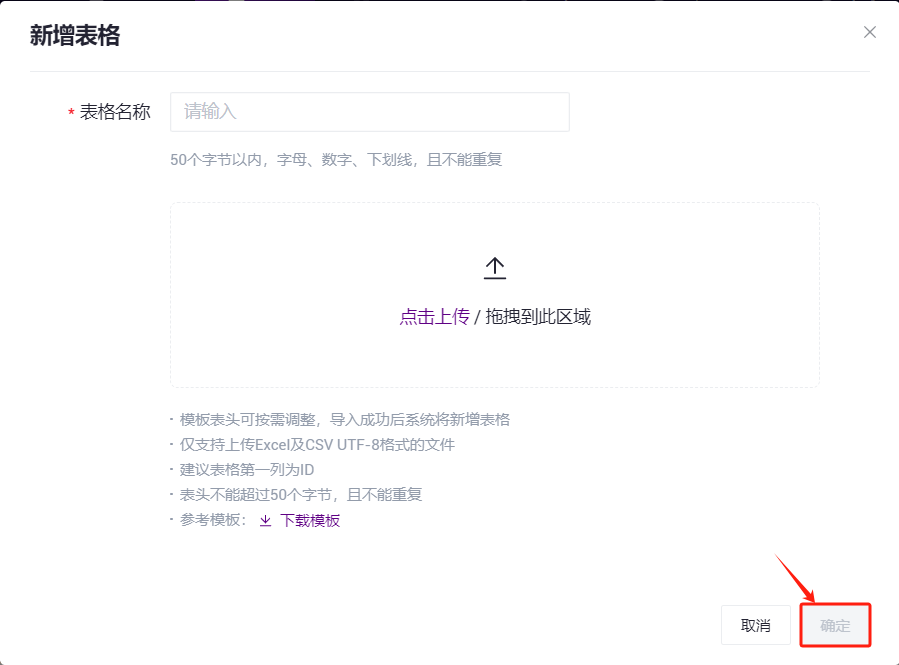
- 点击【点击上传 / 拖拽到此处】浏览并选择已填好样本信息的表格,点击【确认】(图3-42),上传完成后文件会在目标文件夹中显示:
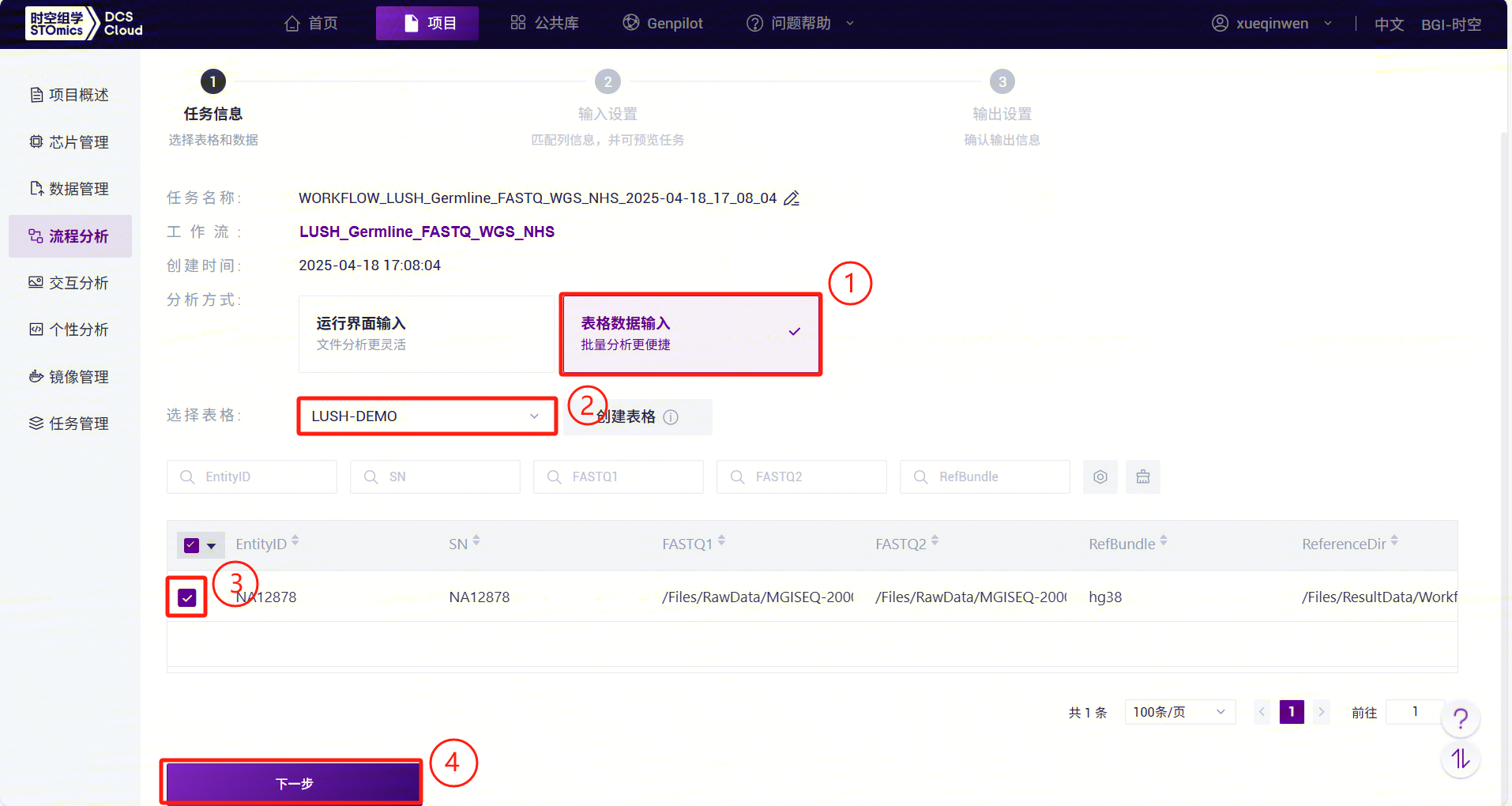
- 点击导航栏【流程分析】进入流程分析页面,在搜索框输入 LUSH_Germline_FASTQ_WGS_NHS ,点击【运行】:
- 选择表格数据输入,点击 请选择表格
 处,选择在本小节3)中导入的表格,选中所需的行,点击【下一步】(图3-43):
处,选择在本小节3)中导入的表格,选中所需的行,点击【下一步】(图3-43):
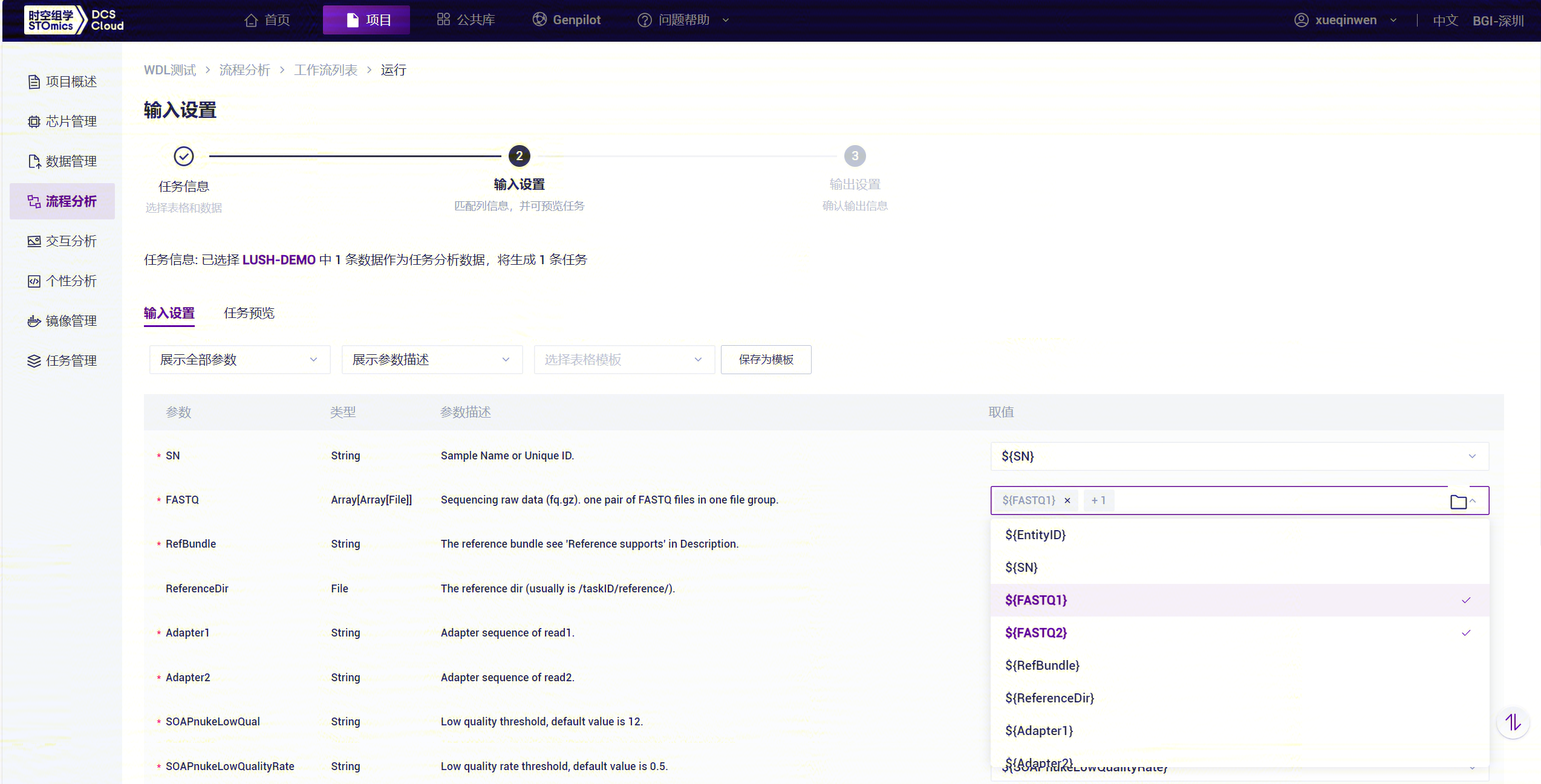
- 在取值处点击并选择对应的值,如FASTQ选取{FASTQ2},注意{FASTQ2}要按顺序选取(如图3-44):
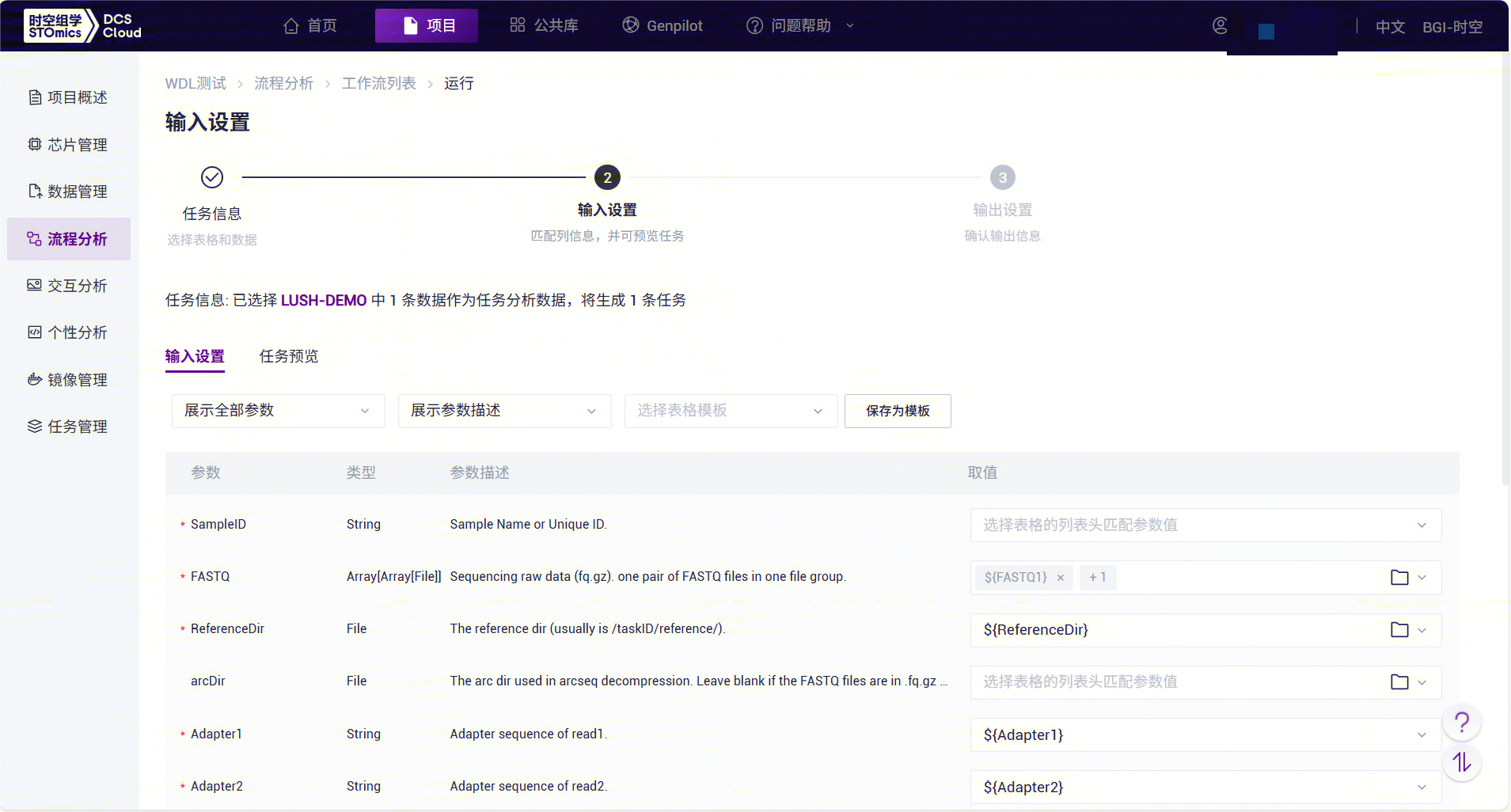
- 录入样本信息,完成后点击【下一步】,确保参数设置无误(如图3-45):
3.7.5 步骤五:启动分析
点击【运行】启动分析(图3-46)
3.8 LUSH结果文件
3.8.1 结果文件下载
- 点击左侧导航栏【任务管理】进入任务管理页面,在流程名称中选择LUSH_Germline_FASTQ_WGS_NHS,查看任务状态,任务完成后,任务状态显示为
 完成,代表任务已完成,即可查看结果部分,此时复制任务编号(图3-47):
完成,代表任务已完成,即可查看结果部分,此时复制任务编号(图3-47):
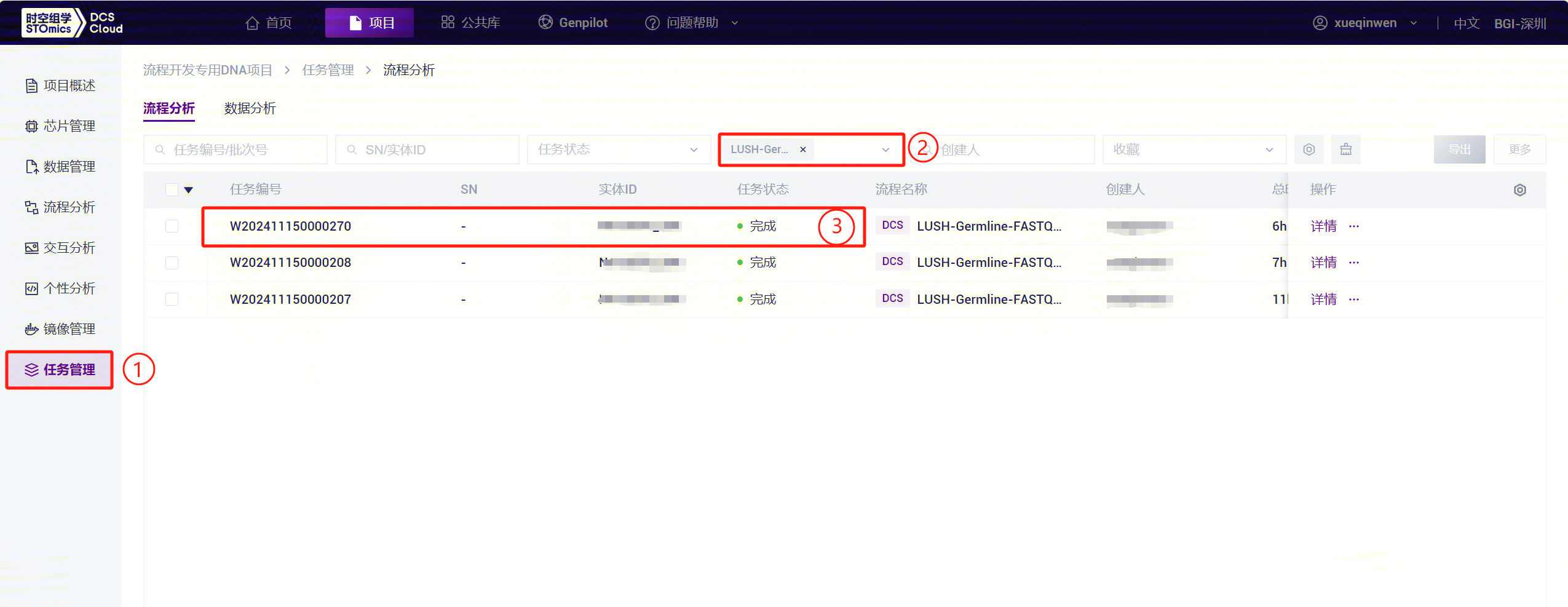
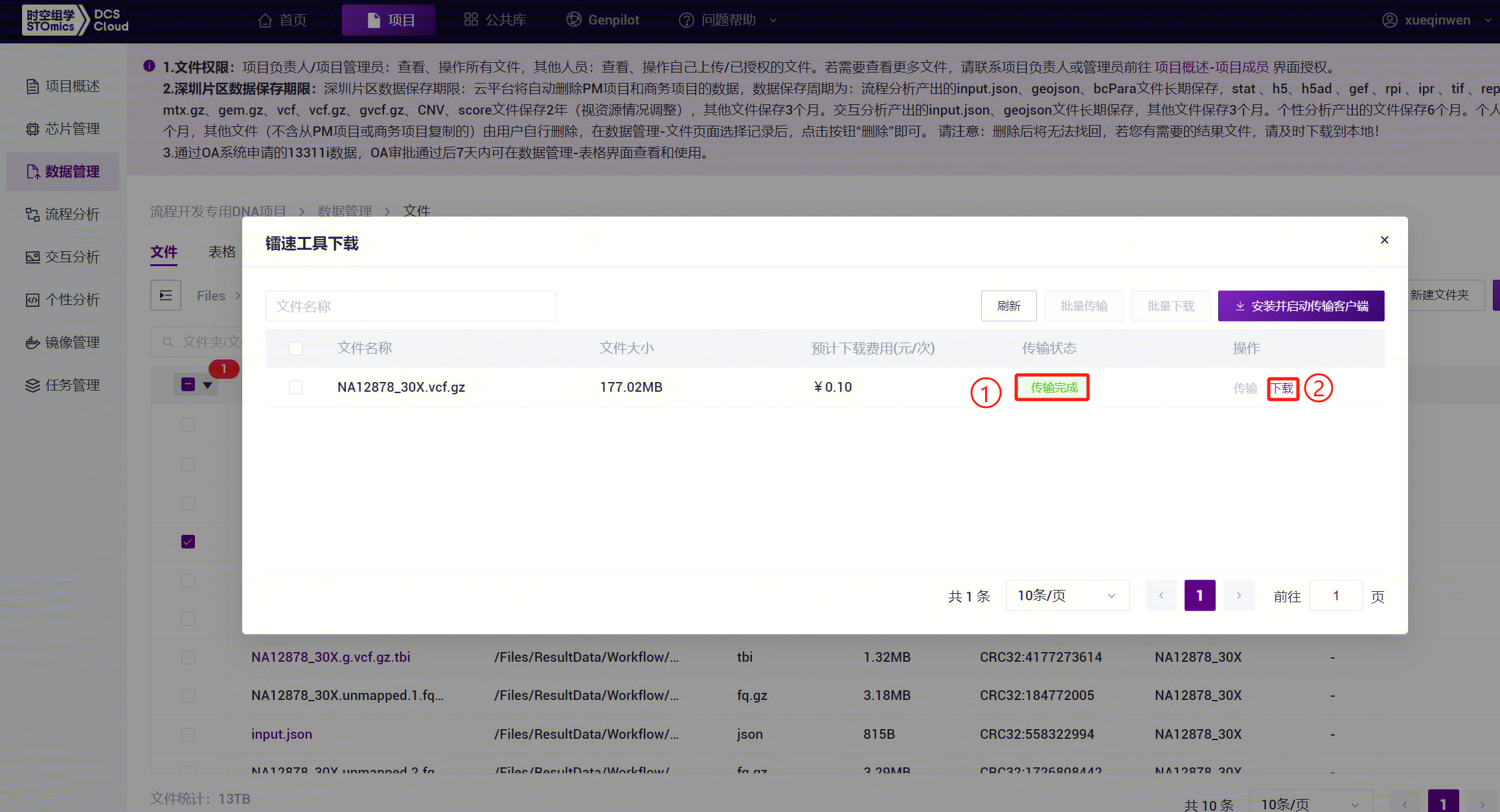
- 选中想要的文件,点击【下载】-【镭速工具下载】(如图3-48):
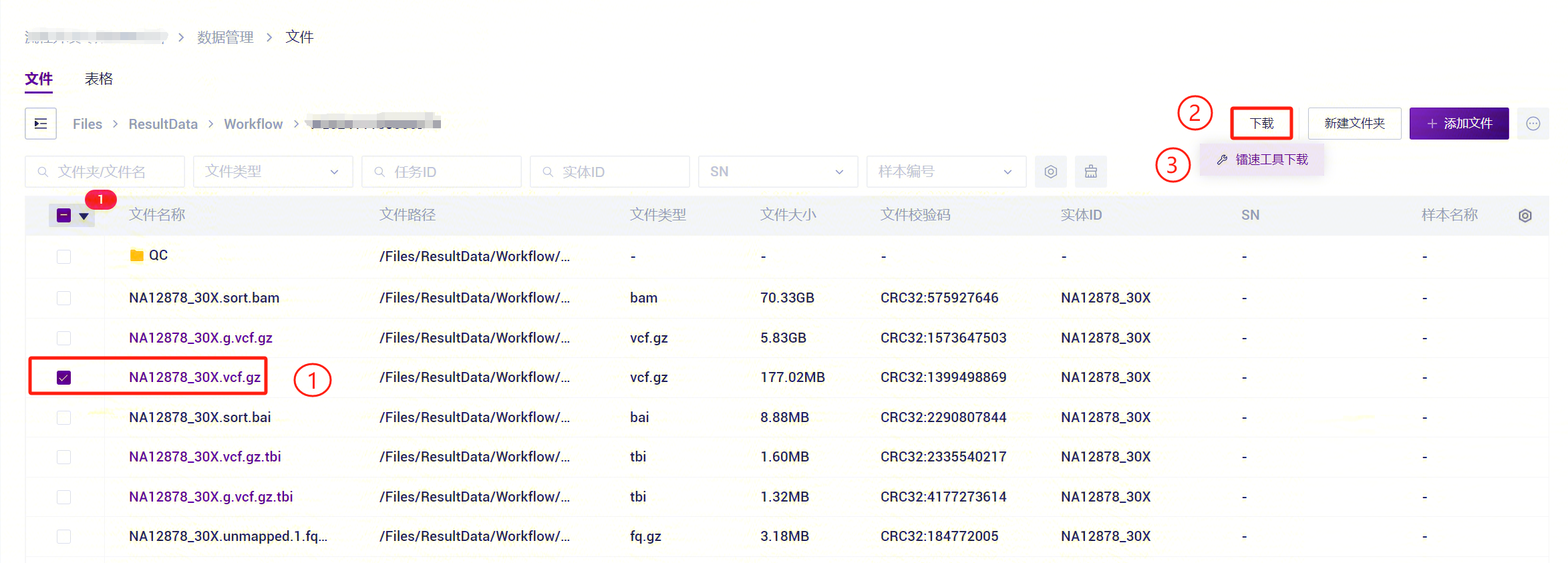
- 在弹出的页面中,点击 传输 :
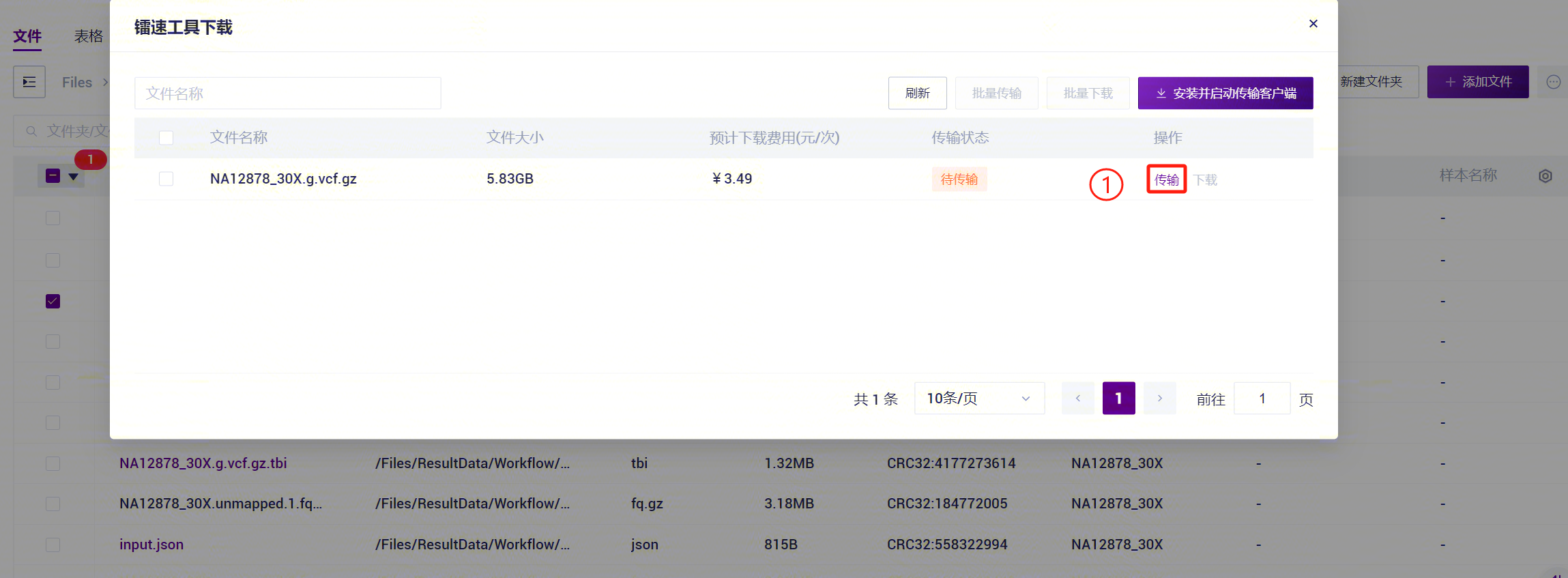
- 待传输状态显示为 传输完成 , 点击 下载,选择文件存放的路径,等待下载完成即可;
3.8.2 LUSH WGS分析流程结果文件
程序运行成功,将在入仓目录下生成如下目录树:
├── sample.genotyper.vcf.gz
├── sample.genotyper.vcf.gz.tbi
├── sample.g.vcf.gz
├── sample.g.vcf.gz.tbi
├── sample.unmapped.1.fq.gz
├── sample.unmapped.2.fq.gz
├── QC
│ ├── Base_distributions_by_read_position_1.txt
│ ├── Base_distributions_by_read_position_2.txt
│ ├── Base_quality_value_distribution_by_read_position_1.txt
│ ├── Base_quality_value_distribution_by_read_position_2.txt
│ ├── Basic_Statistics_of_Sequencing_Quality.txt
│ ├── Distribution_of_Q20_Q30_bases_by_read_position_1.txt
│ ├── Distribution_of_Q20_Q30_bases_by_read_position_2.txt
│ ├── output_filter_files_report.txt
│ └── Statistics_of_Filtered_Reads.txt
└── report
├── sample.AT.xls
├── sample.bamstat.xls
├── sample.base.png
├── sample.cumuPlot.png
├── sample.fqstat.xls
├── sample.histPlot.png
├── sample.insertsize.png
├── sample.qual.png
├── sample\_report\_cn.html
├── sample\_report\_en.html
└── sample.vcfstat.xls
目录说明如下:
| sample.*.vcf.gz* | 变异检测结果和index。 |
|---|---|
| sample.unmapped.*.fq.gz | 未成功比对至参考基因组的Reads文件。 |
| QC | 质控过滤结果输出。 |
| report | 流程结果报告。 |
| report/sample _ report__en.html | 英文版样本分析报告。 |
| report/sample _ report__cn.html | 中文版样本分析报告。 |
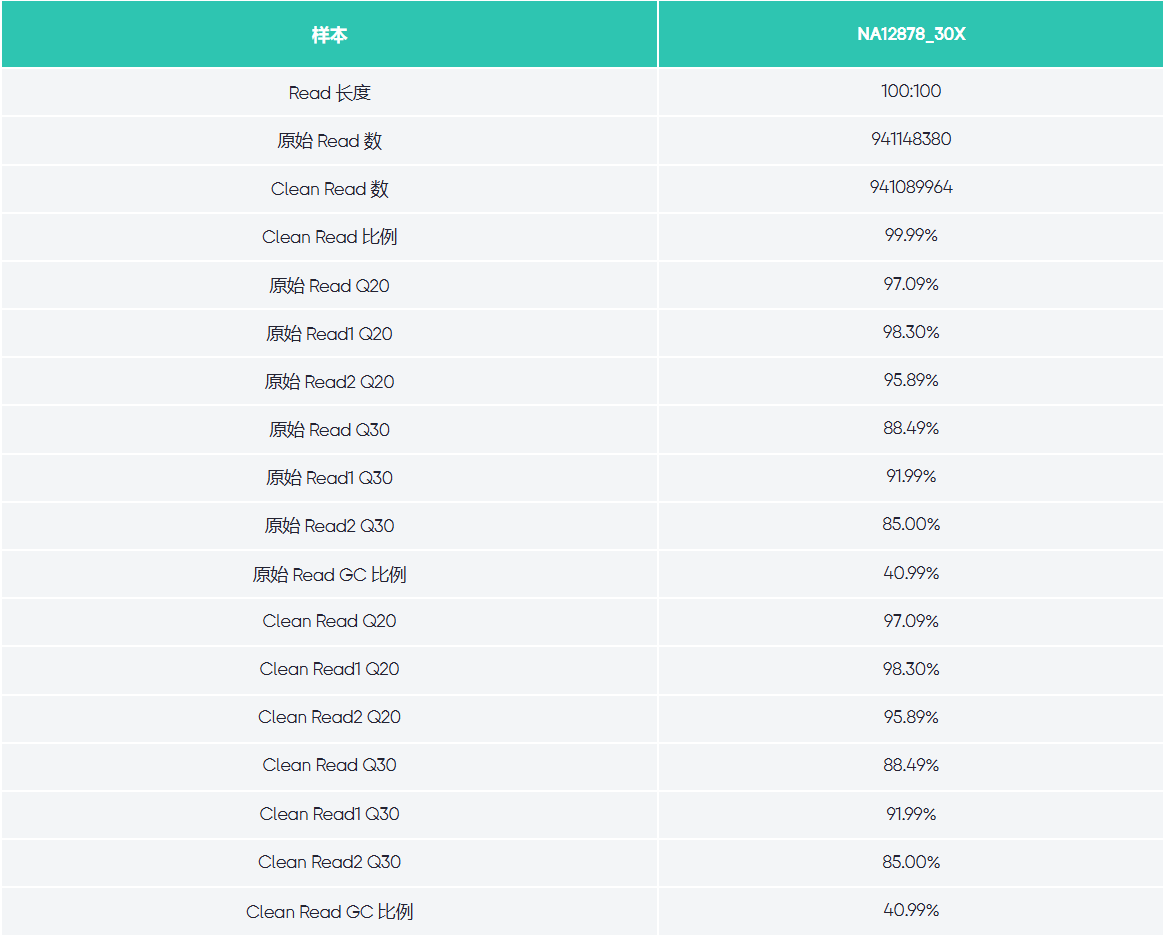
| report/sample.fqstat.xls | 质控结果统计信息 |
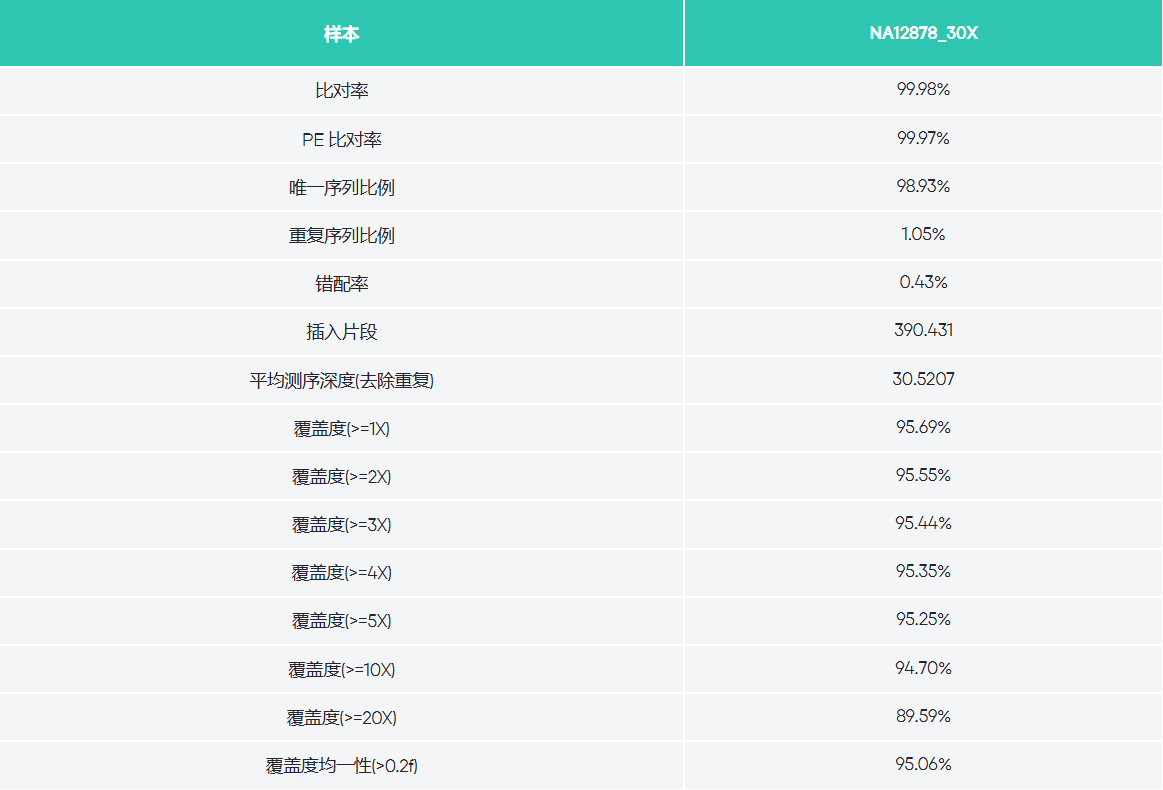
| report/sample. bamstat.xls | 比对结果统计信息 |
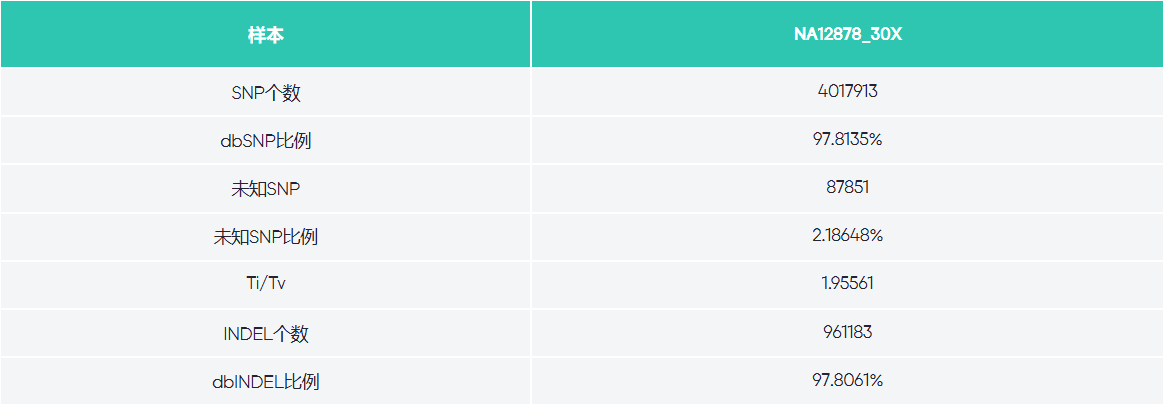
| report/sample.vcfstat.xls | 变异结果统计信息 |
| report/*.png | 报告用图 |
统计结果展示
质控结果统计信息:
比对结果统计信息:
变异结果统计信息:
3.8.3 ExpansionHunter_WGS_STR分析流程结果文件
程序运行成功,将在入仓目录下生成如下目录树:
└── STR
├── sample.json
├── sample\_realigned.bam
└── sample.vcf
目录说明如下:
| sample.json | 有关样本参数和按位点汇总的分析结果的信息的json文件。 |
|---|---|
| sample_realigned.bam | 包含了重新对齐后的读取数据的bam文件。 |
| sample.vcf | 包含STR的位置和信息的VCF文件。 |
3.8.4 PanGenie_WGS_SV分析流程结果文件
程序运行成功,将在入仓目录下生成如下目录树:
├── circos.png
├── circos.svg
├── sample_AnnotSV
│ └── sample-pangenie-genotyping-biallelic-filtered-sv.annotated.tsv
├── sample-pangenie-genotyping-biallelic-filtered-sv.vcf.gz
├──sample-pangenie-genotyping-biallelic-filtered-sv.vcf.gz.tbi
├── pangenie-biallelic
│ ├── sample-pangenie-genotyping-biallelic.vcf.gz
│ └── sample-pangenie-genotyping-biallelic.vcf.gz.tbi
├── pangenie-results_genotyping.vcf
├── pangenie-results_histogram.histo
└── sv_type_counts.txt
目录说明如下:
| sample.*.vcf.gz* | 变异检测结果和index,-filtered为仅包含SV信息的变异检测结果。 |
|---|---|
| sample-pangenie-genotyping-biallelic-filtered-sv.annotated.tsv | 包含SV分型和注释信息结果文件。 |
| pangenie-results_genotyping.vcf | 泛基因组格式的SV变异检测结果。 |
| sv_type_counts.txt | SV统计信息。 |
| *.png *.svg *. histo | 统计用图及画图数据。 |
3.8.5 CNVpytor_WGS_CNV分析流程结果文件
程序运行成功,将在入仓目录下生成如下目录树:
cnv_results/
├── cnv_type_counts.txt
├── sample.CNV.call.txt
├── sample.CNV.manhattan.global.0000.png
├── sample.CNV.rdstat.stat.0000.png
└── out.pytor
目录说明如下:
| cnv_type_counts.txt | CNV统计信息 |
|---|---|
| sample.CNV.call.txt | 包含CNV信息结果文件。 |
| sample.CNV.manhattan.*.png | 曼哈顿热图。 |
| sample.CNV.rdstat.stat.*.png | RD(测序深度)分析统计图。 |
| out.pytor | cnvpytor 文件 (HDF5 文件) |