DCS Tools 用户手册
1. 介绍
1.1 描述
DCS Tools 是一款依托 CPU 加速技术的先进软件,在数据分析上旨在成为 BWA-MEM、GATK 以及大人群联合调用的理想替代方案,在存储上通过压缩Fastq.gz文件大小来减少存储成本。功能包含胚系突变(Germline)和体细胞突变(Somatic)的全基因组(WGS)与全外显子(WES)测序数据分析,joint calling 群体变异检测工具,专门用于基因组数据的FASTQ与VCF文件压缩和解压缩工具。
1.2 优势和价值
1)一致且可靠的变异检测工具
该工具在保证SOAPnuke、BWA和GATK各个模块算法主体逻辑不变的情况下,进行了优秀的并行化设计与重构,加速了计算和减少了不必要的I/O操作。在极大加速的基础上,还保证了结果与gatk体系的高度一致性。
工具亮点:
与 Broad Institute 的 BWA-GATK 最佳实践工作流程采用相同的数学方法,但 FASTQ 到 VCF 的速度快 16倍,胚系的BAM 到 VCF 的速度快 42 倍,体细胞BAM到VCF的速度快8倍(以核·时作为单位进行比较)
无运行间差异,高覆盖区域无下采样
基于 CPU 的系统上运行的纯软件解决方案,跨平台兼容X86、ARM与云环境
阿里云 ecs.i4g.8xlarge
- 30x 基因组,NA12878 (HG001),1.82 小时内(58 个核·时)
2)Joint calling,用于群体测序项目的 gVCF 合并和joint calling工具
支持百万样本的分析,是一种基于Spark的可扩展joint calling软件,已在105,000 个全基因组上应用。
工具亮点
快速:6天完成5.2万人全基因组变异联合检测(joint calling)
准确:GIAB标准品SNP F1值99.55%,INDEL F1值98.95%
规模大:支持百万级别样本的joint calling
易于使用:自动分配任务,无需用户设计并行策略。
3)FASTQ格式数据的压缩工具
沿袭了多数工具的逻辑,将FASTQ中的ID、序列、质量值三部分独立进行压缩,其中序列的压缩除了熵编码,还提供了基于HASH或BWT的序列比对模块,以提高比对成功的序列的压缩率。此外,还使用了数据块的设计,以达到压缩率与性能的平衡,而封装格式的引入,使得数据安全、部分数据独立访问、向下兼容都能轻松完成。
专门优化
一对Pair-end文件可以压缩成单个文件,且压缩率高于单独压缩;
参考基因组序列可以大于4G;
可以对质量值采用有损压缩。
方便灵活
支持Linux、Mac OSX平台,可灵活设置并发线程数,解压无license限制。
生态完整
与DCS Tools中的工具无缝对接,实现边解边算。
数据由基本单元封装,方便进行格式扩展,并保证向下兼容。
4)VCF格式数据的压缩工具
能对大规模基因组测序项目产生的VCF(Variant Call Format)文件进行无损压缩的工具,工具在保证高压缩率的同时,实现了快速的随机访问。将vcf.gz文件压缩至原文件大小的2/5,存储节省60%。
1.3 平台要求
DCS Tools 设计为运行在Linux系统的可执行程序。推荐的Linux发行版为:Centos 7, Debian 8, Ubuntu 14.04及以上的版本。
DCS Tools 对于系统硬件要求如下:cpu 至少有8核,mem至少有128G,并且建议使用读写速度更快的SSD硬盘。
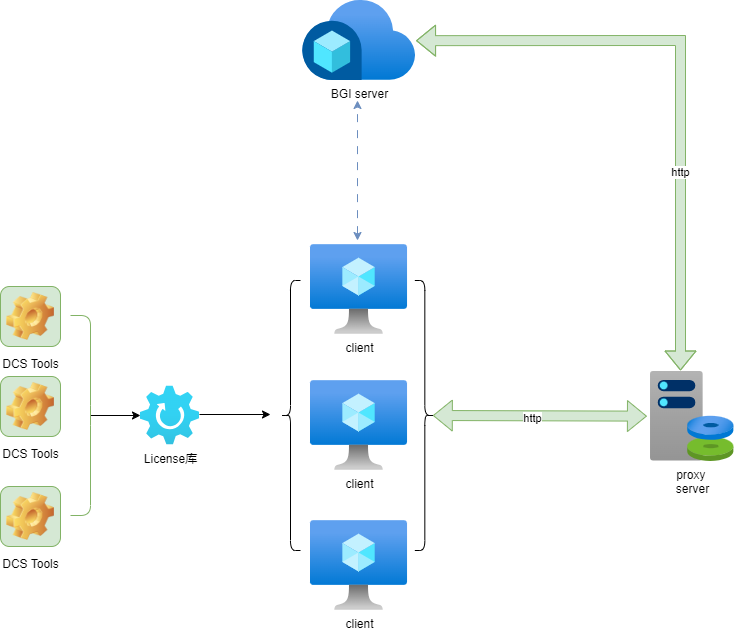
1.4 软件部署(在线认证)
本软件运行时会进行License验证,以确保合法性,在线认证需要一台可以联网(用来与验证服务端通信)的机器,其它机器(往往是计算节点)不需要联网,保证能与联网机器通信即可。当然,单台联网机器也可以部署本软件。认证逻辑可参考本小节末示意图。除了软件包,还需要一份License文件,该文件可以通过云平台(cloud@stomics.tech)或者销售经理获取。
1.4.1 获取联网节点出口IP地址
请保证联网节点不经过额外的HTTP_PROXY或HTTPS_PROXY就能访问cloud.stomics.tech
在申请 License 文件时要求填写联网节点出口 IP 地址,请通过如下命令获取联网节点的出口 IP 地址:
curl --noproxy "*" -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
返回结果中 data 指向所需的 IP 地址:
{"code":0,"msg":"Success","data":"172.19.29.215"}
如果您遇到了如下错误:
curl: (35) error:0A000152:SSL routines::unsafe legacy renegotiation disabled
请改用下面的命令行获取 IP 地址:
echo -e "openssl_conf = openssl_init\n[openssl_init]\nssl_conf = ssl_sect\n[ssl_sect]\nsystem_default = system_default_sect\n[system_default_sect]\nOptions = UnsafeLegacyRenegotiation" | OPENSSL_CONF=/dev/stdin curl --noproxy "*" -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
1.4.2 获取联网节点 MAC 地址
在申请 License 文件时要求填写 MAC 地址,请通过如下命令获取联网节点的 MAC 地址:
./libexec/licproxy --print-mac
1.4.3 获取License文件
向云平台(cloud@stomics.tech)申请或者通过销售经理获取。
1.4.4 获取软件包
从云平台获取程序压缩包并解压,目录 libexec 下包含 License 代理程序(licproxy)。
1.4.5 在联网节点部署 License Proxy
请先在联网节点上使用如下命令在前台运行 License 代理程序:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic
请用您的 License 文件路径代替 /path/to/XXXXXXXXX.lic,该 License 代理程序默认监听 0.0.0.0:8909,如果该地址已被其它程序使用中,您将看到类似警告:
[2025-03-27T06:34:46Z WARN pingora_core::listeners::l4] 0.0.0.0:8909 is in use, will try again
程序将自动重试若干次直到返回,建议您手动 Ctrl + C 停止它后通过添加参数设置其它地址:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic --bind 0.0.0.0:8910
若看到以下输出而没有其它异常,则代表启动成功。
[2025-03-27T07:15:52Z INFO pingora_core::server] Bootstrap starting
[2025-03-27T07:15:52Z INFO pingora_core::server] Bootstrap done
[2025-03-27T07:15:52Z INFO pingora_core::server] Server starting
在进行下一步之前,您可以先通过如下命令检测 License 代理程序和云平台之间的连接是否成功:
curl http://127.0.0.1:8909/api/licensor/licenses/anon/pool
预期看到类似以下输出,代表连接成功:
{"code":0,"msg":"Success"}
在前台运行测试正常后请?Ctrl?+?C?停止它,您可以通过添加选项-d在后台运行License代理程序:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic -d
它将作为守护进程或常驻进程运行。
建议一个节点只需要一个 License 代理在后台运行,也支持同一名 Linux 用户在同一个节点的后台运行多个 License 代理,但不支持多名 Linux 用户在同一个节点的后台运行多个 License 代理,除非您亲自 kill 进程或删除文件 /tmp/pingora.pid
1.4.6 运行dcs工具
如前所述,在联网节点或者能跟联网节点通信的其它节点上运行dcs工具。如何使用本工具集分析WGS或者群体等数据,可以参照quick_start包里的示例脚本。
运行工具前,需要设置好PROXY_HOST环境变量。其值为联网机器的ip和端口,需保证在当前节点可以ping通该ip。如果计算节点和联网节点为同一台机器,ip可以设置为127.0.0.1。
测试计算节点与联网节点的网络
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
运行dcs工具示例:压缩一份fastq数据
export PROXY_HOST=<ip>:<port>
$DCS_HOME/bin/dcs SeqArc compress -i input.fastq.gz -o output.fastq
1.4.7 软件更新后的部署
拿到更新后的软件包后,除非特殊说明,一般不用重新部署license proxy(1.4.4),只需要解压新的软件包,将DCS_HOME设置为新的软件路径即可。
1.5 软件部署(离线认证)
本软件提供离线认证的选项,服务器无需联网即可实现认证,满足医院等单位服务器无法连接互联网时的认证需求。
1.5.1 获取软件和USB加密锁设备
从云平台获取程序压缩包并解压,联系客户支持人员获取USB加密锁设备。
1.5.2 开启非root用户使用USB加密锁设备的权限
使用root权限执行如下命令,开启非root用户使用USB加密锁设备的权限,执行完成后需要重新连接USB加密锁设备使权限生效。
sudo tee /etc/udev/rules.d/ft.rules > /dev/null << EOF
SUBSYSTEMS=="usb",ATTRS{idVendor}=="096e",ATTRS{idProduct}=="0209",MODE="0666"
SUBSYSTEMS=="usb",ATTRS{idVendor}=="096e",ATTRS{idProduct}=="020a",MODE="0666"
EOF
1.5.3 运行dcs工具
连接USB加密锁
将USB加密锁插入到计算机的USB接口。
设置环境变量DCS_DONGLE=1开启离线认证。
export DCS_DONGLE=1检查USB加密锁的用量和到期时间信息。
$DCS_HOME/bin/dcs lickit --query运行成功将返回类似如下信息:
Dongle Information: HID: 46E2005B86013106 PID: B2561B76 UID: 00000000 Is Mother: No DCS Dongle Version: 1.1.0 DCS Dongle Key Created At: 20250904162925 Usage Limits: aligner Limit: 10 variantCaller Limit: 10 jointcaller Limit: 100 seqarc Limit: 10995116277760 bytes (10240.00 G) vararc Limit: 10995116277760 bytes (10240.00 G) alnarc Limit: 10995116277760 bytes (10240.00 G) Current Usage: aligner Usage: 3 variantCaller Usage: 2 jointcaller Usage: 40 seqarc Usage: 98782070 bytes (0.09 G) vararc Usage: 0 bytes (0.00 G) alnarc Usage: 0 bytes (0.00 G) Deadline: 2025-12-31 23:59:59显示加密锁的基本信息,各个工具的用量限制,当前用量和到期时间。
运行dcs工具示例:压缩一份fastq数据
export DCS_DONGLE=1 $DCS_HOME/bin/dcs SeqArc compress -i input.fastq.gz -o output.fastq注意事项
离线认证模式下,dcstools使用文件锁进行同进程同步以确保用量更新准确,文件锁的路径是/tmp/dcstool_dongle.lock。计算jointcaller用量时,如果分区间并行执行jointcaller,例如同一批输入gvcf文件,分chr1,chr2,...染色体区间执行dcs jointcaller任务,为了避免重复计算样本量,需要记录历史jointcalling样本信息,仅第一次执行jointcaller增加样本用量。dcstools默认将jointcaller运行的历史记录保存在/home/$USER/.local/share/dcstools/jointcaller_history.dat文件中,设置DCS_CONFIG_DIR环境变量可以更改这个路径,例如export DCS_CONFIG_DIR=/mnt/disk/dcstools/,历史记录将保存到/mnt/disk/dcstools/jointcaller_history.dat。
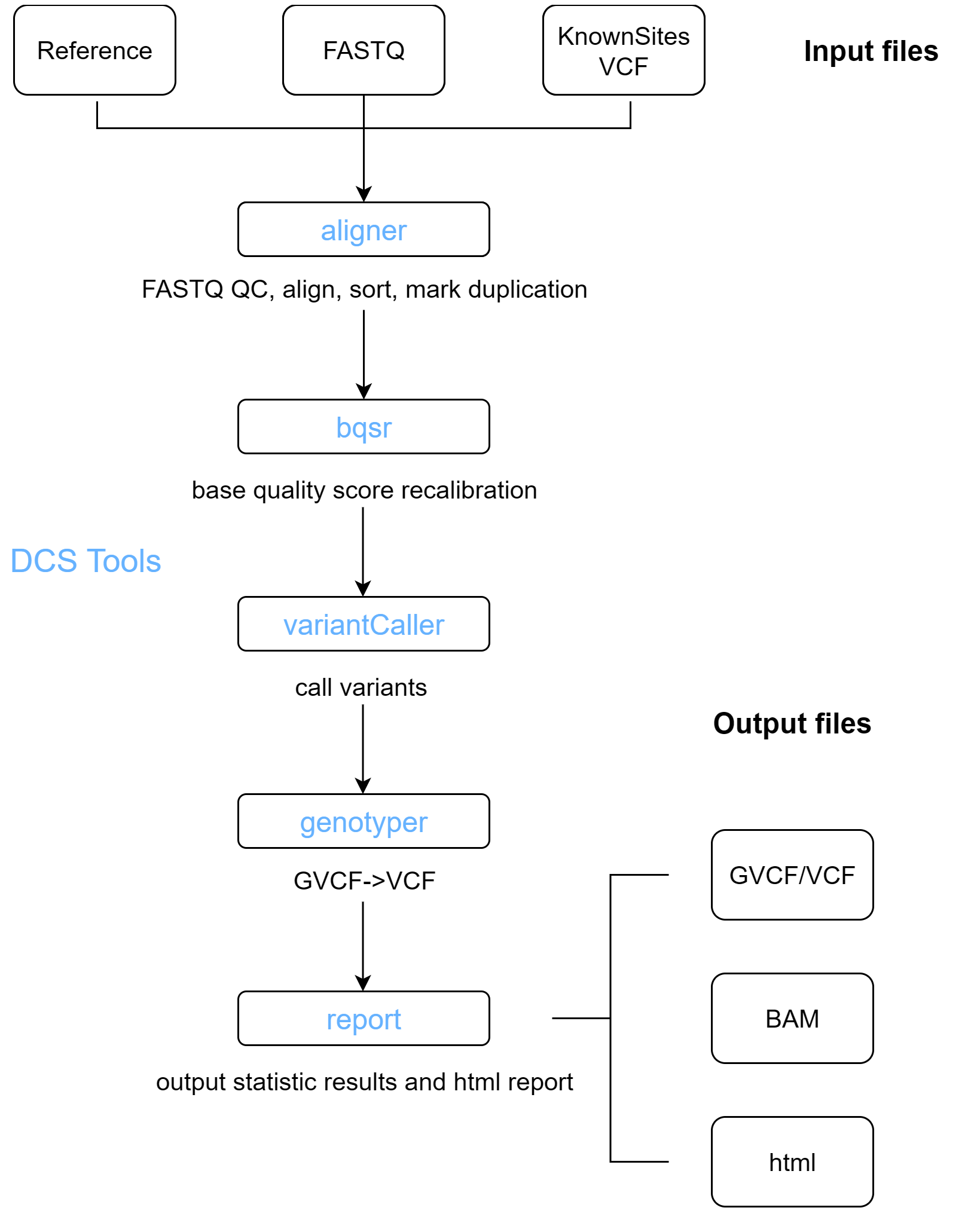
1.6 工具列表
| 场景 | 工具(模块)名称 | 功能 |
|---|---|---|
| WGS/WES分析 | aligner | 质控、比对、排序和标记重复 |
| bqsr | 碱基质量值矫正 | |
| variantCaller | 变异检测(类似GATK haplotypeCaller) | |
| genotyper | GVCF->VCF | |
| report | 输出报告 | |
| 群体基因组联合分析 | jointcaller | 联合变异检测,支持百万级样本 |
| FASTQ数据压缩解压 | SeqArc | 高效无损FASTQ压缩工具 |
| VCF数据压缩解压 | VarArc | 高效无损VCF压缩工具 |
| 体细胞变异检测 | Mutect2 | 体细胞变异检测 |
| license查询等 | lickit | 查询可用线程数和测试网络连接 |
| 小工具 | extract-vcf | 简化vcf文件,缩小文件大小,减轻IO压力,主要针对bqsr和genotyper的knownSites(dbsnp等)输入 |
| bam-stat | 统计比对信息 | |
| vcf-stat | 统计变异信息 | |
| tbi2lix | 将tbi索引文件简化为自定义的线性索引文件,用于jointcalling |
2. 典型用法
2.1 WGS
WGS(Whole Genome Sequencing, 全基因组测序)是一种能够测定生物体全部基因组序列的技术。WGS分析流程一般包括数据质控、序列比对、标记重复、碱基质量值重校正、变异检测等步骤。DCS tools 遵循原始算法,对算法实现进行了性能优化,优化后的分析流程比BWA-GATK快16倍。
2.1.1 准备工作
WGS分析流程的输入文件如下:
- 序列比对工具需要的文件;
| 索引文件 | 描述 |
|---|---|
| species.fa | 参考基因组FASTA文件 |
| species.fa.fai | 用于快速定位和随机访问FASTA文件的序列区域 |
| species.dict | 索引作用,为后续处理流程提供参考序列的快速查找表 |
| species.fa.* | 比对索引文件 |
通过aligner工具构建比对索引,索引文件放置在FASTA同级目录,比对索引构建命令是:
dcs aligner --threads <INT> --build-index <FASTA>
- FASTQ测序数据,支持gz格式;
- 单核苷酸多态性数据库(dbSNP)数据和已知变异位点文件,以VCF格式提供;
DCS tools 工具包提供的WGS分析流程主要包括以下步骤:
- 序列比对和标记重复:在序列进行比对之前会进行质控,将经过质控的序列比对到FASTA文件中记录的参考基因组上,比对结果在进行标记重复后写入BAM文件;
- 碱基质量值重校正:调整分配给序列每个碱基的质量分数,消除因测序方法引入的实验偏差;
- 变异检测:识别出测序数据相对参考基因组存在的变异位点,并为每个样本在这些位点计算基因型;
2.1.2 序列比对和标记重复
将序列比对到参考基因组上确定每条序列的位置,为进一步检测变异提供基础;标记重复消除PCR扩增产生的重复序列,降低假阳性提高变异检测准确性,这些步骤由软件alinger完成。而且aligner 还集成了数据质控功能,可以在序列比对之前对序列进行过滤和处理,从而降低错误率,避免后续分析受到噪音数据的干扰。
dcs aligner --threads THREADNUM \
--aln-index INDEX \
--fq1 FASTQ1 \
--fq2 FASTQ2 \
--read-group RGROUP \
--bam-out BAMFILE \
--fastq-qc-dir QCDIR \
--high-speed-storage TEMPDIR
该命令需要以下输入:
- THREADNUM: 线程数量,建议该值不要超过CPU数量;
- INDEX: 参考基因组FASTA文件路径,在FASTA同级目录下存放比对软件构建的索引文件;
- FASTQ1: PE测序的FASTQ1文件路径;
- FASTQ2: PE 测试的FASTQ2文件路径;
- RGROUP: 格式是“@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM“, 其中GROUP_NAME 是read group标识符,SAMPLE_NAME表示样品名称,PLATFORM表示测序平台;
- BAMFILE: 经过标记重复后的BMA文件;
- QCDIR: 质控报告的输出位置;
- TEMPDIR: 临时文件的存储路径,若该路径处于高速存储设备,则有利于提升软件性能;
2.1.3 碱基质量值矫正
由于存在系统误差,序列碱基质量值并不总是完全准确,需要对序列的碱基质量值重校正。该步骤可以纠正系统偏差生成更精确的质量分数,提高变异检测准确性,减少假阳性和假阴性。统一质量评估标准符合GATK推荐的最佳实践,该步骤由软件bqsr完成。
dcs bqsr --threads THREADNUM \
--in-bam BAMFILE \
--reference FASTA \
--known-sites KNOWN_SITES \
--recal-table RECAL_TABLE
该命令需要以下输入:
- THREADNUM: 线程数量,建议该值不要超过CPU核数;
- BAMFILE: 输入文件,序列比对和标记重复后生成的BAM文件;
- FASTA: 参考基因组FASTA文件路径,确保使用参考基因组和序列比对阶段使用的相同;
- KNOWN_SITES: 单核苷酸多态性数据库数据和已知变异位点文件;
- RECAL_TABLE: 输出文件,保存用来矫正碱基质量值的信息
2.1.4 变异检测
检测SNP和INDEL变异,生成gvcf文件,同时生成bam的统计信息,统计文件与GVCF同目录,名字为bamStat.txt。该步骤由软件variantcaller完成
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-t THREADUM
该命令需要以下输入:
- INPUT_BAM:2.1.2输出的BAM文件(排序标记重复后)。
- OUTPUT_GVCF:输出gvcf文件。
- REFERENCE:参考基因组FASTA文件。
- RECAL_TABLE:输入文件,包含矫正碱基质量值所需要的信息,由2.1.3步骤生成。
- THREADNUM:线程数量,建议该值不要超过CPU核数。
对于单个样本,如果仅仅需要获取包含变异记录的vcf文件,可以使用genotyper软件。
dcs genotyper -I INPUT_GVCF \
-O OUTPUT_VCF \
-s QUAL \
-t THREDNUM
该命令需要以下输入:
- INPUT_GVCF:来自于variantcaller的GVCF文件。
- OUTPUT_VCF:输出VCF文件。
- QUAL:变异质量值阈值,大质量值大于或等于QUAL的记录将被输出。
- THREADNUM:线程数量,建议该值不要超过CPU核数。
2.1.5 报告生成
可以根据每个样本的统计文件进行整合,生成可视化HTML报告文件
dcs report --sample SAMPLE_NAME \
--filter QCDIR \
--bam_stat BAMStat \
--vcf_stat VCFStat \
--output OUTPUT_DIR
该命令需要以下输入:
- SAMPLE_NAME:样本命名。
- QCDIR:aligner生成的质控统计报告文件夹。
- BAMStat:variantCaller结果目录下的bamStat.txt。
- VCFStat:vcf统计结果,默认在工作目录下的vcfStat.txt。
- OUTPUT_DIR: 报告输出目录。
2.2 WES
与WGS步骤基本相同,仅在variantCaller步骤有区别,该步骤需要增加一个区间参数(-L)
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-L INTERVAL_FILE
-t THREADUM
INTERVAL_FILE:记录WES区间的bed文件,按照惯例其中每行格式如下
chr<tab>start<tab>end
其中start和end位置是0-based表示,属于左闭右开的区间。
2.3 Joint Calling
Joint Calling是一种检测群体变异的方法,这里特指以多个gvcf文件为输入,合并多个样本的变异并重新计算样本基因型的过程。DPGT是一个分布式的spark程序,可以高效完成大规模样本的Joint Calling工作。dcs jointcaller是DPGT工具的一个包装器,用于运行单机模式的DPGT。
# 设置JAVA_HOME为jdk8
export JAVA_HOME=/path_to/jdk8
# 将spark bin加入到PATH,用于调用spark-submit
export PATH=/path_to/spark/bin/:$PATH
dcs jointcaller \
-i GVCF_LIST \
-r REFERENCE \
-o OUTDIR \
-j JOBS \
--memory MEMORY \
-l REGION
DPGT依赖java8,请设置JAVA_HOME=/path_to/jdk8。spark推荐2.4.5及以上的版本,spark 2.4.5下载路径:https://archive.apache.org/dist/spark/spark-2.4.5/spark-2.4.5-bin-hadoop2.6.tgz
该命令需要如下输入:
GVCF_LIST:输入的gvcf列表,是一个文本文件,每行一个gvcf路径。注意gvcf需要有匹配的tbi索引文件,程序默认通过gvcf路径+.tbi查找gvcf相匹配的tbi索引文件。
REFERENCE:参考基因组fasta文件路径,注意这个fasta文件需要与生成gvcf是使用的fasta文件一致。程序同时需要与fasta文件匹配的.dict和.fai文件,例如fasta文件名是hg38.fasta,那么程序也需要读取hg38.dict, hg38.fasta.fai。
OUTDIR:输出文件夹,结果vcf路径是OUTDIR/result.*.vcf.gz。
JOBS:并行的任务数目,使用的线程数目是JOBS个,JOBS是一个整型值,范围是1到机器的线程数目。
MEMORY:java最大堆内存大小,内存的单位是"g"或"m"。内存大小和JOBS值和样本量存在关联,对于1000样本,推荐每个job分配2g内存,对于2000-5000样本,推荐每个job分配4g内存,对于5000-100000样本,推荐每个job分配6g内存,对于100000-500000样本,推荐每个job分配8g内存。
REGION:目标区域。支持输入字符串和一个bed路径,区域字符串格式与bcftools定义的格式一致,例如:"chr20:1000-2000", "chr20:1000-", "chr20",如果需要指定多个区域,请指定多个-l参数,例如指定处理chr1和chr2:1000-2000可以这样做:-l chr1 -l chr2:1000-2000
2.4 SeqArc(FASTQ压缩)
SeqArc 模块用于对 FASTQ数据进行高效压缩与解压缩。该模块针对二代短读长和三代长读长测序数据分别优化了压缩策略,并支持三种不同的压缩模式:
- 无参考模式 — 不依赖任何参考文件,压缩速度较快,压缩比则较低。
- 单参考模式 — 使用指定的参考基因组(FASTA),在压缩/解压过程中提高压缩比与一致性。
- 多参考模式 — 提供多个参考基因组,程序自动匹配最合适的单个FASTA用于压缩与解压。
相比通用压缩工具,SeqArc模块在存储效率和解压速度上具有显著优势,尤其适用于大规模测序数据存档和传输场景。
2.4.1 环境与输入输出要求
2.4.1.1 待压缩文件要求
FASTQ 文件,支持未压缩文本格式(.fq或.fastq)及gz压缩格式(.fq.gz或.fastq.gz)。
2.4.1.2 参考文件要求
如果使用单参考模式或多参考模式,需要提供参考基因组 FASTA 文件。
若使用多参考模式,索引目录中应有多个 FASTA 文件。
需要下载并解压 NCBI taxonomy 数据包 taxdump.tar.gz(http://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz)至该目录(占485MB空间),以支持参考基因组匹配。
2.4.1.3 压缩输出文件格式
压缩后:.arc 文件。
解压后:FASTQ或FASTQ.gz 文件(gz可指定压缩级别)。
2.4.2 索引构建
2.4.2.1 用法(Usage)
dcs SeqArc index [options] -r /path/to/ref.fa
dcs SeqArc index [options] -K /path/to/ref_dir/
2.4.2.2 功能说明
建立参考基因组的索引,以支撑“单参考压缩”与“多参考压缩”。
如果指定 -K(多个参考 FASTA 所在目录),将构建分类数据库,使压缩阶段能自动选择最合适的参考,解压阶段也能自动选择压缩时使用的参考。
用「dcstools SeqArc index -r REF -o OUTDIR 」命令会生成两个用于压缩的索引,分别是二代短读长的索引和三代长读长的索引。以人类基因组为例,GRCh38.fa为3.1GB,建索引耗时约半小时,输出二代短读长索引GRCh38.fa.mini_28_10,占14GB,输出三代长读长索引GRCh38.fa.similar_26_23,占6.1GB,++过程中峰值内存约35GB,对于比人类基因组更大的FASTA,消耗内存会成比例增加++。
2.4.2.3 参数说明
| 参数 | 说明 | 默认值 / 要点 |
|---|---|---|
-r, --ref <file> | 指定一个参考 FASTA 文件 | 必要用于单参考模式 |
-K, --clsdb <dir> | 指定包含多个参考 FASTA 的目录 | 用于物种识别模式 |
-H, --hitlimit <int> | 在参考匹配时最大的匹配数 | 默认 2 |
-t, --thread <int> | 使用线程数 | 默认 8 |
-o, --output <dir> | 索引输出目录 | 若未指定则使用当前目录或默认子目录 |
-h, --help | 显示帮助信息 | — |
2.4.3 压缩
2.4.3.1 用法(Usage)
dcs SeqArc compress [options] -i reads.fq -o reads.arc
dcs SeqArc compress [options] -r /path/to/ref.fa -i reads.fq -o reads.arc
dcs SeqArc compress [options] -K /path/to/ref_dir/ -i reads.fq -o reads.arc
2.4.3.2 功能说明
将 FASTQ 文件压缩为 .arc 格式。
三种压缩模式可选:无参考、单参考、多参考。
值得注意的是,尽管多参考模式使用了多个FASTA,但只会从多个建库的FASTA中尝试找到与当前FASTQ最接近的一个(或者因皆不匹配,不使用任何一个,选择无参考模式),在压缩和解压过程中依然只有一个FASTA文件作为参考。
2.4.3.3 参数说明
| 参数 | 说明 | 默认值 / 建议 |
|---|---|---|
-i, --input <file> | 输入 FASTQ 文件 | 必填 |
-o, --output <file> | 输出文件,会增加判定,保证输出文件后缀为 .fq.arc或.fastq.arc | 必填 |
-r, --ref <file> | 单参考 FASTA | 可选 |
-K, --clsdb <dir> | 多参考目录,用于物种识别 | 可选 |
-H, --hitlimit <int> | 参考匹配数上限 | 默认 2 |
-J, --clsmin <float> | 最小读 hit 比例 | 默认 0.6 |
-L, --clsnum <int> | 用于分类判断的读数样本数目 | 默认 10000 |
-t, --thread <int> | 线程数 | 默认 8 |
-f, --force | 覆盖已有输出文件 | — |
--pipein | 从管道中读取输入 | 适合流式处理 |
--inputname <str> | 指定原始FASTQ 名称,仅当使用 --pipein 时有用,该名称于解压且不指定输出文件名时被使用 | — |
--blocksz <size> | 块大小,以 MB 为单位 | 默认 50 |
--ccheck | 压缩后立即解压校验内容,慢但安全 | — |
--calcmd5 | 计算输入 FASTQ 的 MD5 校验和 | 用于后续校验 |
--slevel <int> | 序列压缩级别(8–15) | 默认 10 |
--verify <int> | 校验模式:0(无),1(整个块),2(整个块 + 名称、序列、质量分别校验) | 默认 1 |
-h, --help | 显示帮助信息 | — |
2.4.4 解压
2.4.4.1 用法(Usage)
dcs SeqArc decompress [options] -i reads.arc -o reads.fq
dcs SeqArc decompress [options] -r /path/to/ref.fa -i reads.arc -o reads.fq
dcs SeqArc decompress [options] -K /path/to/ref_dir/ -i reads.arc -o reads.fq
2.4.4.2 功能说明
将 .arc 文件解压为 FASTQ 文件。
如果压缩时用了参考或物种识别库,则解压时也应提供相应参考或参考库,以保证正确恢复序列。
2.4.4.3 参数说明
| 参数 | 说明 | 默认值 / 建议 |
|---|---|---|
-i, --input <file> | 输入 .arc 文件 | 必填 |
-o, --output <file> | 输出 FASTQ 文件路径(若不指定则输出到标准输出) | — |
-r, --ref <path> | 单参考 FASTA 文件或目录 | 如果压缩时使用了 -r 模式 |
-K, --clsdb <dir> | 多参考目录,用于物种识别 | 如果压缩时使用了 -K 模式 |
-t, --thread <int> | 线程数 | 默认 8 |
-f, --force | 覆盖已有输出文件 | — |
--type <int> | 输出类型:0 = gz 压缩 FASTQ;1 = 纯 fastq 文本 | 默认 0 |
--gzlevel <int> | gzip 压缩级别 | 默认 4 |
--dcheck | 写入后再次读入并校验 | 较慢但更可靠 |
--show | 若压缩时使用了 --calcmd5,显示原始 FASTQ 的 MD5 值 | — |
-h, --help | 显示帮助信息 | — |
2.4.5 检查
2.4.5.1 用法(Usage)
dcs SeqArc check [options] -i reads.arc
dcs SeqArc check [options] -r /path/to/ref.fa -i reads.arc
dcs SeqArc check [options] -K /path/to/ref_dir/ -i reads.arc
2.4.5.2 功能说明
检查 .arc 文件是否完全无损。
如果压缩时用了参考或物种识别库,则检查时也应提供相应参考或参考库。
2.4.5.3 参数说明
| 参数 | 说明 | 默认值 / 建议 |
|---|---|---|
-i, --input <file> | 输入 .arc 文件 | 必填 |
-r, --ref <path> | 单参考 FASTA 文件或目录 | 如果压缩时使用了 -r 模式 |
-K, --clsdb <dir> | 多参考目录,用于物种识别 | 如果压缩时使用了 -K 模式 |
-t, --thread <int> | 线程数 | 默认 8 |
-f, --force | 覆盖已有输出文件 | — |
-h, --help | 显示帮助信息 | — |
2.4.6 输出统计信息
2.4.6.1 用法(Usage)
dcs SeqArc stat [options] -i reads.arc
2.4.6.2 功能说明
输出 .arc 文件在压缩过程中统计得到的数据。
2.4.6.3 参数说明
| 参数 | 说明 | 默认值 / 建议 |
|---|---|---|
-i, --input <file> | 输入 .arc 文件 | 必填 |
-h, --help | 显示帮助信息 | — |
2.4.7 参数选择与使用建议
| 状况 | 建议模式 | 关键参数调优 |
|---|---|---|
| 数据量非常大,希望最大压缩率 | 单参考 (-r) 或 多参考 (-K) 模式 | 提高 --slevel,使用更多线程;确认参考基因组与样本物种匹配良好 |
| 数据种类混杂、物种未知 | 多参考模式 | 准备好多个参考 FASTA,并下载 taxonomy 数据;调节 -L、-clsmin 确保分类准确 |
| 要求速度快,对压缩率要求不高 | 无参考模式 | 使用默认压缩级别即可,不用 -r 或 -K;减少校验选项(如关闭 --ccheck、--verify) |
| 希望压缩与解压后的数据完全一致 | 开启校验选项 | 用 --ccheck(压缩后校验),用 --dcheck(解压校验),保存 MD5 校验值 |
2.4.8 示例
2.4.8.1 索引构建
首先需要进行文件系统的句柄设置
# 二者选一即可 # 仅对当前会话临时生效,若需要长期生效,需添加至环境变量 ulimit -n 65535 # 设置软限制为65535 ulimit -Hn 65535 # 设置硬限制为65535(需要root权限)对于单参考的情况,直接创建参考序列的索引文件
dcs SeqArc index -r REF -o OUTDIR对于多参考的情况,创建物种识别库索引和每个物种的索引文件
# i.新建一个多物种文件目录
mkdir fasidx && cd fasidx
# ii.下载物种关系文件并解压
wget http://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
tar xzf taxdump.tar.gz
# iii.下载需要用到的各个物种FASTA到/path/to/fa目录中,将fa.gz解压成fa
gunzip ./*fa.gz
# iv.创建物种识别库索引clsf.cdb,clsf_del.cdb,clsf_opt.cdb,clsf_tax.cdb
dcs SeqArc index -K .
# v.为每个物种(如aaa.fa)创建对应的比对索引
dcs SeqArc index -r aaa.fa
2.4.8.2 使用dcs进行压缩
用户如果知道fastq的物种,可以使用【-r】参数直接选择对应物种的参考序列进行压缩:
dcs SeqArc compress -r REF \ -i FASTQ \ -o ARC \ -t THREADNUM如果用户不知道具体物种,可以使用物种识别库进行有参压缩,程序会利用物种识别库分析输入数据的物种,然后选用对应的索引文件进行有参压缩
dcs SeqArc compress -K REFDIR \ -i FASTQ \ -o ARC \ -t THREADNUM如果用户没有参考序列,也可以进行无参压缩
dcs SeqArc compress -i FASTQ \
-o ARC \
-t THREADNUM
该命令需要输入下述参数:
- REF: 输入的参考序列路径
- REFDIR :包含多物种参考序列的目录
- FASTQ:输入的fastq文件路径
- ARC: 输出的压缩文件路径
- THREADNUM: 设置的线程数
2.4.8.3 使用dcs进行解压
用户如果知道压缩使用的具体参考序列,可以使用【-r】参数直接选择对应物种的参考序列进行压缩
dcs SeqArc decompress -r REF \ -i ARC \ -o FASTQ \ -t THREADNUM用户如果使用物种识别库进行的压缩,解压时需要指定物种识别库路径
dcs SeqArc decompress -r REFDIR \ -i ARC \ -o FASTQ \ -t THREADNUM用户如果是无参压缩,解压也是无参解压
dcs SeqArc decompress -i ARC \
-o FASTQ \
-t THREADNUM
该命令需要输入下述参数:
- REF: 输入的参考序列路径
- REFDIR :包含多物种参考序列的目录
- FASTQ:输出的fastq文件路径
- ARC:输入的压缩文件路径
- THREADNUM: 设置的线程数
2.4.8.4 使用单独解压工具进行解压
如果客户没有购买DCS Tools,单需要解压缩arc格式的文件,可以用独立的免费解压工具对数据进行解压缩,解压工具软件包(Seqarc_decompress)向销售获取。
用户如果知道压缩使用的具体参考序列,可以使用【-r】参数直接选择对应物种的参考序列进行压缩
SeqArc_decompress -r REF \ -i ARC \ -o FASTQ \ -t THREADNUM用户如果使用物种识别库进行的压缩,解压时需要指定物种识别库路径
SeqArc_decompress -r REFDIR \ -i ARC \ -o FASTQ \ -t THREADNUM用户如果是无参压缩,解压也是无参解压
SeqArc_decompress -i ARC \
-o FASTQ \
-t THREADNUM
该命令需要输入下述参数:
- REF: 输入的参考序列路径
- REFDIR :包含多物种参考序列的目录
- FASTQ:输出的fastq文件路径
- ARC:输入的压缩文件路径
- THREADNUM: 设置的线程数
2.4.9 注意事项
- 若压缩时使用
-r或-K模式,解压时必须提供相应参考或目录,以保证内容完整且一致性高。 - 多线程可以加速,但资源(CPU + 内存)受限时可能引起性能瓶颈。
- 较高压缩级别(
--slevel大)与校验选项会显著增加运行时间。 - 文件覆盖(
-f, --force)慎用,以免误覆盖重要数据。 - 在使用
--pipein或从标准输入读取时,推荐手动指定--inputname用于正确标识输入文件名,避免解压后文件名不明确。
2.5 VarArc
VarArc 压缩是将普通的 VCF 文本或者 VCF.gz 文件压缩成更小的文件进行替代,以节省存储空间,减少数据传输时间,提高处理效率。
压缩命令如下:
dcs VarArc compress -i VCF \
-o ARC \
-t THREADNUM \
--rowcnt RCNT \
--blkcolcnt BCNT
该命令需要输入下述参数:
- VCF: 输入的vcf文件路径
- ARC :输出的压缩文件路径
- THREADNUM: 设置的线程数
- RCNT :设置行数,每RCNT为一个大块
- BCNT:设置样品列数,每BCNT为一个小块
解压命令如下:
dcs VarArc decompress -i ARC \
-o VCF \
-t THREADNUM \
-p POSRANGE \
-s SAMPLELIST
该命令需要输入下述参数:
- ARC: 输入的压缩文件路径
- VCF :输出的解压vcf文件路径
- THREADNUM: 设置的线程数
- POSRANGE :设置查询的染色体名称和区间 【chr1: 1000-2000】
- SAMPLELIST:设置查询的样品命令列表文件【文件的每一行是一个样品】
view可以解压查看vcf内容但是不生成文件,命令如下:
dcs VarArc view -i ARC \
-t THREADNUM \
-p POSRANGE \
-s SAMPLELIST
该命令需要输入下述参数:
- ARC: 输入的压缩文件路径
- THREADNUM: 设置的线程数
- POSRANGE :设置查询的染色体名称和区间 【chr1: 1000-2000】
- SAMPLELIST:设置查询的样品命令列表文件【文件的每一行是一个样品】
3. 工具参数详细说明
3.1 aligner
aligner支持的命令行参数如下:
- --threads: 线程数量,建议不超过CPU核数
- --fq1: 输入数据fastq1; 若是多文件,支持逗号分割,支持文件列表;
- --fq2:输入数据fastq1; 若是多文件,支持逗号分割,支持文件列表;
- --seqarc-ref:SeqArc格式文件的解压需要的索引文件;
- --read-group: 格式是“@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM“, 其中GROUP_NAME 是read group标识符,SAMPLE_NAME表示样品名称,PLATFORM表示测序平台;
- --build-index: 构建比对工具需要的索引文件;
- --aln-index:序列比对需要的FASTA路径,注意配套的索引文件和FASTA处于相同目录;
- --mark-split-hits-secondary:将较短的拆分命中标记为次要
- --use-soft-clipping-for-sup-align:使用软裁剪进行补充比对
- --no-markdup:不进行标记重复;
- --bam-out:输出BAM文件
- --high-speed-storage:临时文件的存储路径,若该路径处于高速存储设备,则有利于提升软件性能;
- --fastq-qc-dir: FASTQ 质控报告输出目录;
- --no-filter:不过滤FASTQ数据
- --output-clean-fq:输出clean reads
- --qualified-quality-threshold: 合格碱基质量阈值(默认是12)
- --low-quality-ratio: 低质量值碱基比例阈值(默认是0.5)
- --mean-qual: 平均质量值阈值
- --quality-offset: 质量值偏移,典型值为33、64。若设置为0,则该值由程序推断(默认值:33)
- --adapter1:read1的接头序列,全大写
- --adapter2:read2的接头序列,全大写
- --adapter-max-mismatch:adapter查找时最大允许碱基错配数量(默认值是2)
- --adapter-match-ratio:adapter的最短匹配长度比例(默认是0.5)
- --trim-adapter:从reads序列中截去adapter, 而不是直接丢弃reads;
- --min-read-len:最小reads长度阈值(默认是30)
- --n-base-ratio:N碱基比例阈值(默认是0.1)
- --trim-ends: 截去reads两端一定数量的碱基,格式是逗号分隔的整数;
3.2 bqsr
bqsr支持的命令行选项如下:
- --threads: 线程数量,建议不超过CPU核数;
- --in-bam: 经过标记重复的BAM文件;
- --out-bam: 碱基质量值校正后生成BAM存放的路径,如果不加该参数,将会只生成recal table;
- --reference: 参考基因组FASTA文件,该文件和序列比对阶段使用的参考基因组相同;
- --recal-table: 输出文件,保存用来矫正碱基质量值的信息
- --known-sites: 单核苷酸多态性数据库数据和已知变异位点文件,支持文本和gz压缩格式;
- --interval: BED文件;
- --interval-padding: BED区间两边填充碱基个数;
3.3 variantCaller
variantCaller的详细参数说明如下:
- -I, --input
<file1><file2>...:输入bam文件路径,可以接受多个bam路径,以空格分割。 - -O, --output
<file>:输出vcf/gvcf BGZF文件,程序自动创建相应的索引文件。 - -R, --reference
<file>:参考基因组fasta文件路径。 - --recal-table
<file>: 输入文件,用来矫正碱基质量值 - -H, --help:打印帮助信息。
- --version:打印版本号。
- -L, --interval
<file>:目标区间bed文件路径。 - -P, --interval-padding
<int>: 区间向两侧扩展的大小,必须是非负整数。(默认值:0) - -Q, --base-quality-score-threshold
<int>:碱基质量值阈值,质量值低于该阈值时将会被置为最小值6。(默认值:18) - -D, --max-reads-depth
<int>:每个比对起始位置保留的最大reads数目。reads数目超过该阈值时将进行下采样。(默认值:50) - -G, --gvcf-gq-bands
<int1><int2>...:参考置信模型(Reference confidence model)GQ分组, 输入空格分隔的多个int。(默认值:[1,2,3,...,59,60,70,80,90,99]) - -t, --threads
<int>:程序使用的线程数目,范围是[1,128]。(默认值:30) - --pcr-indel-model
<str>: PCR indel模型,参数范围:{NONE, HOSTILE, CONSERVATIVE, AGGRESSIVE}。(默认值:CONSERVATIVE) - --emit-ref-confidence
<str>:运行参考置信打分模型,参数范围:{NONE, BP_RESOLUTION, GVCF},NONE:不运行参考置信打分模型;BP_RESOLUTION:运行参考置信打分模型,每个位点都输出一个变异记录;GVCF:运行参考置信打分模型,按照-G参数对参考置信位点(ALT=<NON_REF>)按照GQ值进行分组输出。(默认值:NONE) - --compression-level
<int>:输出vcf/gvcf BGZF文件压缩等级,范围是[0,9]。(默认值:6)
3.4 genotyper
genotyper的功能是处理单个样本的gvcf文件,输出仅仅包含变异记录的vcf文件,如果需要对处理多个gvcf文件并进行联合变异检测(joint calling)请使用高性能的DPGT软件。genotyper的详细参数说明如下:
- -I, --input
<file>: 输入gvcf文件路径。 - -O, --output
<file>:输出包含基因型的vcf BGZF文件,程序自动创建相应的索引文件。 - -s, --stand-call-conf
<float>: 变异质量值阈值,质量值大于或等于该值的变异将被输出。(默认值10.0) - -D, --dbsnp
<file>: dbsnp文件 - -t, --threads
<int>:程序使用的线程数目。(默认值6) - --version:打印版本号。
- -h, --help:打印帮助信息。
3.5 jointcaller (DPGT)
jointcaller是DPGT工具的一个包装器,用于便捷地运行单机模式DPGT。DPGT是一个分布式Joint Calling工具,它的详细参数说明如下:
- -i,--input
<FILE>:输入gvcf文件列表,是一个文本文件,每行一个gvcf路径。. - --indices
<FILE>:可选参数,程序默认通过在gvcf路径末尾加.tbi获取索引文件路径,通过该参数可以指定索引文件路径,该参数需要输入一个文本文件,每行一个索引路径,索引路径的顺序需要与gvcf路径的顺序匹配。. - -use-lix
<Boolean>:使用lix索引格式。lix文件是DPGT实现的一种简化索引文件,可以通过tbi2lix程序将tbi索引转换成lix索引,lix索引的大小仅仅是tbi索引的约1/80。可选参数:true, false(默认值:false) - --header
<FILE>:可选参数,使用一个预先计算的vcf头文件,避免重复计算。 - -o,--output
<DIR>:输出文件夹。 - -r,--reference
<FILE>:参考基因组fasta文件。 - -l,--target-regions
<STRING>:目标区域字符串或bed文件,默认处理全基因组。 - -j,--jobs
<INT>:并行任务数目 (默认值:10) - -n,--num-combine-partitions
<INT>:合并变异阶段的分区数目,默认值是样本数目的平方根,向下取整。(默认值:-1) - -w,--window
<INT>:每个合并-基因型计算过程处理的区域大小,(默认值:300M) - -d,--delete
<Boolean>:是否删除临时文件。可选参数: true, false,(默认值:true) - -s,--stand-call-conf
<FLOAT>:质量值阈值,变异记录的质量值大于或等于该值时将被输出,(默认值:30.0) - --dbsnp
<FILE>:dbsnp vcf文件路径,用于注释dbsnp ID。 - --use-old-qual-calculator
<Boolean>:是否使用旧质量值计算器,设为true时使用旧质量值计算器,旧质量值计算器的结果与GATK v4.1.0设置--use-old-qual-calculator true --use-new-qual-calculator false结果匹配,设为false时使用新质量值计算器,新质量值计算器的结果与GATK v4.1.1后的版本结果匹配,可选参数: true, false,(默认值:true) - --heterozygosity
<FLOAT>:杂合SNP变异的先验概率,(默认值:0.001) - --indel-heterozygosity
<FLOAT>:杂合INDEL变异的先验概率,(默认值:1.25E-4) - --heterozygosity-stdev
<FLOAT>:SNP和INDEL变异先验概率的标准差, (默认值:0.01) - --max-alternate-alleles
<INT>:最大等位基因数目,等位基因数目超过该值的变异,程序仅保留可能性最大的该值个等位基因,(默认值:6) - --ploidy
<INT>:倍性, (默认值:2) - -h,--help:打印帮助信息并退出。
- -V,--version:打印版本号并退出。
3.6 SeqArc
3.6.1 创建索引功能, 子命令【index】
创建索引参数如下:
- -h, --help :获取创建索引参数使用帮助信息
- -r, --ref
<file>: 设置输入的参考序列文件(fasta)路径 - -K, --clsdb
<dir>: 设置输入的多个物种的参考序列文件所在的目录,目录下面需要存在文names.dmp和nodes.dmp, 这两个文件可以从ttp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz 下载 - -H, --hitlimit
<int>: 设置在分类法上minimizer的命中计数,默认是【2】 - -t, --thread
<int>: 设置线程数,默认是【8】 - -o, --output
<dir>: 设置输出目录,用于存放生成的参考序列文件对应的索引文件 - --aligntype
<int>: 设置生成索引文件的比对方式,默认是【2】 【1--hash 2--minimizer 3-- longseq】
已知fastq的物种,可以使用【-r】参数直接选择对应物种的参考序列生成对应的索引文件,使用【-o】参数保存生成的索引文件,然后使用这个索引文件进行压缩解压。
未知fastq的物种,可以使用【-K】参数对目录下所有的参考序列创建一个物种识别库索引。然后再对每一个参考序列使用【-r】参数创建对应的索引文件,此时【-o】的目录需要和【-K】的输入目录保持一致。
3.6.2 压缩功能, 子命令【compress】
压缩参数如下:
- -h, --help : 获取压缩参数的使用帮助说明
- -r, --ref
<file>: 设置输入的参考序列路径,该路径下需要存在对应生成的索引文件 - -K, --clsdb
<dir>:设置输入的多个物种的参考序列文件所在的目录,该目录下需要存在生成的物种识别库文件,和各个物种对应的索引文件 - -H, --hitlimit
<int>: 设置在分类法上最小化器的命中计数,默认是【2】。保持和创建参数一致 - -J, --clsmin
<float>:设置物种识别时,判断物种的最小命中率,默认是【0.6】 - -L, --clsnum
<int>:设置物种识别时,需要从fastq读取多少条read用于判别种类,默认是【10000】 - -t, --thread
<int>: 设置线程数,默认是【8】 - -f, --force : 设置输出文件是否强制覆盖已有的同名文件
- -i, --input
<file>: 设置输入的fastq文件 - --pipein : 设置输入的是否是管道数据
- -o, --output
<file>:设置输出的文件路径,会自动在尾部添加 【.arc】后缀 - --blocksz
<int>:设置压缩时每个数据块大小,默认是【50】 - --ccheck : 设置是否在压缩完成且数据落盘后,再执行一遍解压逻辑,保证数据的完整性
- --calcmd5 :设置是否在压缩时计算输入文件的md5,并保存到压缩文件中
- --aligntype
<int>:设置选用的压缩比对方式,默认是【2】 - -a, --archive
<dir>: 设置输入的要打包压缩的目录,可对目录下的文件进行压缩后打包 - --slevel
<int>: 设置seq的压缩阶乘,默认是【10】 - --verify
<int>: 设置每个数据块的校验方式,默认是【1】【0:不校验 1:校验整个数据块 2:分别校验name,seq,qual】
3.6.3 解压功能, 子命令【decompress】
解压参数如下:
- -h, --help : 获取解压参数的使用帮助说明
- -r, --ref
<dir/file>: 设置输入的具体参考序列路径,该路径下需要存在对应生成的索引文件。或者输入的物种识别库目录,该目录下需要存在各个物种的索引文件。 - -t, --thread
<int>: 设置线程数,默认是【8】 - -f, --force : 设置输出文件是否强制覆盖已有的同名文件
- -i, --input
<file>: 设置输入的压缩结果文件路径 - -o, --output
<file>: 设置输出的解压文件路径 - --rawtype : 设置解压输出文件和压缩原始文件格式保持一致,只对mgz格式文件生效
- --dcheck : 设置解压完成且数据落盘后,再读取解压落盘数据块,并对块进行校验,保证解压的完整性
- -x, --extract : 提取压缩包的一个或者多个文件,需要输入文件是【-a, --archive】生成的打包文件
- -l, --list : 显示压缩包内的文件信息,需要输入文件是【-a, --archive】生成的打包文件
- --show : 显示压缩文件的文本md5值,需要压缩时指定【--calcmd5】
3.7 VarArc
3.7.1 压缩功能, 子命令【compress】
- -h, --help : 获取压缩参数的使用帮助说明
- -i, --input
<file>: 设置输入的vcf文件 - -o, --output
<file>:设置输出的压缩文件路径 - -t, --thread
<int>: 设置线程数,默认是【4】 - --rowcnt :设置每个大块的行数
- --blkcolcnt: 设置每个小块的样品数
3.7.2 解压压功能, 子命令【decompress】
- -h, --help : 获取压缩参数的使用帮助说明
- -i, --input
<file>: 设置输入的压缩文件 - -o, --output
<file>:设置输出的解压文件路径 - -t, --thread
<int>: 设置线程数,默认是【4】 - -p, --pos: 设置筛选的染色体名称和范围 【chr1: 1000-2000】
- -s, --samplefile: 设置筛选的样品名称列表文件,每个样品名称占用一行
- -S, --samplelist: 设置筛选的样品名称列表,样品名称之间用逗号分隔
3.7.3 查看功能, 子命令【view】
- -h, --help : 获取压缩参数的使用帮助说明
- -i, --input
<file>: 设置输入的压缩文件 - -t, --thread
<int>: 设置线程数,默认是【4】 - -p, --pos: 设置筛选的染色体名称和范围 【chr1: 1000-2000】
- -s, --samplefile: 设置筛选的样品名称列表文件,每个样品名称占用一行
- -S, --samplelist: 设置筛选的样品名称列表,样品名称之间用逗号分隔
3.8 license-proxy
可执行文件 license-proxy 参数如下:
- -h, --help:打印帮助信息
- --license
<LICENSE_PATH>:License 文件路径 - --bind
<BIND_ADDR>:监听地址 - -d, --daemon:是否在后台启动
3.9 report
对WGS/WES分析结果统计文件进行整理,画图,并生成HTML格式的报告。参数如下:
- --sample : 样本名称
- --filter : 过滤目录路径
- --bam_stat : BAM统计文件路径
- --vcf_stat : VCF统计文件路径
- --output : 输出报告的目录
- --is_se : (可选)如果使用单端测序,加上该标志。
3.10 lickit
用于检查网络的联通性,以及查询用户可用的线程数。
- --ping
<ip:port>: 检查网络是否联通 - --query
<ip:port>: 查询用户当前可用的线程数 - --uplog
<ip:port logpath [msg]>:用户主动上传问题日志
3.11 小工具(Utility tools)
3.11.1 extract-vcf
常规vcf文件每行含有8列字段,其中6,7,8列内容占用空间大但是程序不需要这些信息,为了减小IO压力,该工具只从vcf文件中提取前5列信息,后3列内容置为*号。主要参数如下:
- --known-sites
<file>: 输入vcf文件,支持文本和gz压缩格式; - --output-mini-vcf
<file>: 输出vcf文件,若输出文件名带有gz, 则为压缩格式;
3.11.2 bam-stat
根据bam文件,统计比对信息,在WGS流程中,该功能已集成到variantCaller中,也可以单独运行。其参数为:
- --bam
<file>: 输入bam文件路径 - --bed
<file>: 目标区间,如果是WGS数据,可以指定非N区文件(如果不方便提供,使用variantCaller的bam统计结果即可)。如果是WES数据,则指定目标区域文件 - --output
<file>: 输出文件路径
3.11.3 vcf-stat
根据genotyper后的vcf文件,统计变异信息,包括snp数量等,其参数为:
- --vcf
<file>: 输入vcf文件路径,支持文本和gz格式
结果默认输出到标准输出,可以重定向到指定文件(参照4.1的step 5)。
3.11.4 tbi2lix
- -i, --input FILE: 输入tbi索引文件
- -o, --output FILE: 输出lix索引文件
- -m, --min-shift INT: 设置lix线性索引的最小区间大小为2^INT (默认值14,对应区间大小16K)
- -h, --help: 打印帮助信息并退出
3.12 Mutect2
该模块为GATK的并行加速版本,用--nthread来设置并行线程数量,用--java-options "<Xmx等参数>"指定java/jvm参数, 其它参数说明可以参考GATK
3.13 fastq2vcf
该自动化脚本($DCS_HOME/libexec/fastq2vcf.sh)为了方便WGS/WES流程搭建,将参数统一传给该脚本,完成从fastq到vcf的全过程。详细参数如下
Usage: fastq2vcf.sh DCS_HOME=/path/to/dcs-tools refFasta=/path/to/ref.fa FQ1=/path/to/read1.fq.gz FQ2=/path/to/read2.fq.gz [other parameters]
Required parameters:
DCS_HOME: path to the dcs-tools directory
refFasta: path to the reference genome fasta file
FQ1: path to the first FASTQ file
Optional parameters:
FQ2: path to the second FASTQ file (for paired-end data)
proxyHost: proxy host for license authentication (default:127.0.0.1:8909)
OUTPUT_DIR: output directory (default: current directory)
threads: number of threads to use (default:32)
sample: sample name (default:test)
bed: path to target regions in bed format (optional)
useLowMemIndex: whether to use low memory index (true/false, default:false)
ploidy: sample ploidy (default:2), if greater than 2, java and spark environment is required for genotype calling
dbsnp: path to dbsnp vcf file (optional)
knownSitesFileList: comma-separated list of known sites vcf files for BQSR (optional)
Example:
fastq2vcf.sh DCS_HOME=/data/dcs-tools refFasta=/data/ref/hg38.fa FQ1=/data/sample_R1.fq.gz FQ2=/data/sample_R2.fq.gz sample=sample1 threads=16 useLowMemIndex=true ploidy=2 knownSitesFileList=/data/known1.vcf,/data/known2.vcf dbsnp=/data/dbsnp.vcf
4. 快速开始——使用示例脚本和数据
按照1.4部署好软件后,获取quick start数据包(从销售经理处获取),解压后修改示例脚本中的DCS_HOME和PROXY_HOST值,DCS_HOME为软件包解压后的路径,PROXY_HOST为联网节点的ip和1.4中部署时指定的端口(默认8909)。不同应用场景执行相应的示例脚本。
tar xzvf dcs-quick-start-xxxx.tar.gz
cd dcs-quick-start
4.1 fastq2vcf
从v2.0开始,DCS Tools工具集中新增了fastq2vcf的自动化脚本,支持WGS和WES数据从fastq到vcf过程的脚本自动化过程。如果要自定义该过程,可以参考4.2和4.3。使用方法以human常规WGS数据示例
$DCS_HOME/libexec/fastq2vcf.sh \
DCS_HOME=<dcs home> \
refFasta=<ref fasta> \
FQ1=<r1.fq/gz> \
FQ2=<r2.fq/gz> \
OUTPUT_DIR=<output dir> \
sample=<sample name> \
dbsnp=<dbsnp vcf> \
knownSitesFileList=<some known sites with vcf format seperated by comma>
4.2 WGS
参考脚本为quick_start_wgs.sh
#!/bin/bash
# This script provides a quick start guide for using the dcs tool to analyze whole-genome sequencing data.
# The script demonstrates how to build the index of the reference genome, align the reads,
# recalibrate the base quality scores, call variants, and filter the variants.
# if you have any questions, please contact us at cloud@stomics.tech
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
if [ ! -d "$DCS_HOME" ]; then
echo "--- Error: DCS_HOME directory does not exist: $DCS_HOME"
exit 1
fi
if [ ! -r "$DCS_HOME" ]; then
echo "--- Error: No read permission for DCS_HOME: $DCS_HOME"
exit 1
fi
# Before using the dcs tool, you must obtain a license file and start the authentication proxy program
# on a node that can connect to the internet.
# Get the local IP address, which is used for license application.
# The output data field of the following command indicates the IP address.
# curl -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
# start the proxy program
# LICENSE=<path/to/license/file>
# $DCS_HOME/bin/dcs licproxy --license $LICENSE
# Set the following environment variables
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
# Set the following variables for the dcs tool
FQ1=$SCRIPT_DIR/data/fastq/simu.r1.fq.gz
FQ2=$SCRIPT_DIR/data/fastq/simu.r2.fq.gz
FA=$SCRIPT_DIR/data/ref/chr20.fa
KN1=$SCRIPT_DIR/data/known_sites/1000G_phase1.snps.high_confidence.hg38.chr20.vcf.gz
KN2=$SCRIPT_DIR/data/known_sites/dbsnp_151.hg38.chr20.vcf.gz
KN3=$SCRIPT_DIR/data/known_sites/hapmap_3.3.hg38.chr20.vcf.gz
KN4=$SCRIPT_DIR/data/known_sites/Mills_and_1000G_gold_standard.indels.hg38.chr20.vcf.gz
nthreads=8
sample=simu
group="@RG\\tID:sample\\tLB:sample\\tSM:sample\\tPL:COMPLETE\\tCN:DCS"
# run the wgs pipeline ------------------------------------------------------------------------------------
# check the existence of the index files
for file in $FA.0123 $FA.amb $FA.ann $FA.bwt $FA.index1 $FA.index2 $FA.pac $FA.sa;
do
if [ ! -f $file ]; then
echo "File $file does not exist, try to build the index ..."
$DCS_HOME/bin/dcs aligner --threads 16 --build-index $FA
break;
fi
done
# Step 0: Set the working directory
workDir=$SCRIPT_DIR/result_wgs
mkdir -p $workDir
mkdir -p $workDir/${sample}.qcdir
# Step 1: Alignment
$DCS_HOME/bin/dcs aligner --threads $nthreads --aln-index $FA --fq1 $FQ1 --fq2 $FQ2 --read-group $group --no-filter --fastq-qc-dir $workDir/${sample}.qcdir --bam-out $workDir/${sample}.align.bam
# Step 2: base quality score recalibration
$DCS_HOME/bin/dcs bqsr --threads $nthreads --in-bam $workDir/${sample}.align.bam --reference $FA --known-sites $KN1 --known-sites $KN2 --known-sites $KN3 --known-sites $KN4 --recal-table $workDir/${sample}.recal.table
# Step 3: variant calling
$DCS_HOME/bin/dcs variantCaller --threads $nthreads --input $workDir/${sample}.align.bam --output $workDir/${sample}.variantCaller.g.vcf.gz --reference $FA --recal-table $workDir/${sample}.recal.table
# Step 4: genotype calling
$DCS_HOME/bin/dcs genotyper --threads $nthreads --input $workDir/${sample}.variantCaller.g.vcf.gz --output $workDir/${sample}.genotyper.vcf.gz --dbsnp $KN2 -s 30.0
# Step 5: generate the final report
$DCS_HOME/bin/dcs vcf-stat --vcf $workDir/${sample}.genotyper.vcf.gz >$workDir/vcfStat.txt
$DCS_HOME/bin/dcs report --sample ${sample} --filter $workDir/${sample}.qcdir --bam_stat $workDir/bamStat.txt --vcf_stat $workDir/vcfStat.txt --output $workDir/report
修改文件中的环境变量后,直接运行即可。
# 修改DCS_HOME和PROXY_HOST值
vi quick_start_wgs.sh
sh quick_start_wgs.sh
多倍体数据分析使用示例
export PATH=$PATH:<your spark-submit directory>
$DCS_HOME/libexec/fastq2vcf.sh DCS_HOME=<dcs home> proxyHost=<ip:port of license proxy> refFasta=<ref.fa> FQ1=<r1.fq/gz> FQ2=<r2.fq/gz> ploidy=<ploidy> OUTPUT_DIR=<output dir>
该脚本的GVCF到VCF步骤使用的是DCS Tools的jointcaller模块,默认与旧版GATK(4.1.5之前)结果一致,如果要拿到与GATK(4.1.5之后)新版一致的结果,需要在fastq2vcf.sh中的jointcaller脚本部分(如下,最后一行),加上这样的参数:--use-old-qual-calculator false
if [ "$useDPGT" = true ]; then
GVCF_FOFN=$workDir/gvcf.fofn
ls $workDir/${sample}.variantCaller.g.vcf.gz > ${GVCF_FOFN}
$DCS_HOME/bin/dcs jointcaller \
--input ${GVCF_FOFN} \
--output ${workDir} \
--reference ${FA} \
--jobs ${nthreads} \
--memory 1G \
--use-old-qual-calculator false
fi
4.3 WES
与WGS步骤基本相同,仅在variantCaller步骤有区别,该步骤需要增加一个区间参数(-L),填上wes区间的bed文件
# Step 3: variant calling
$DCS_HOME/bin/dcs variantCaller --threads $nthreads --input $workDir/${sample}.align.bam --output $workDir/${sample}.variantCaller.g.vcf.gz --reference $FA --recal-table $workDir/${sample}.recal.table -L <wes.target.bed>
4.4 Joint Calling
参考脚本为quick_start_jointcalling.sh,与WGS类似,修改其中的DCS_HOME和PROXY_HOST环境变量,此外dcs jointcaller需要设置JAVA_HOME为jdk8,并将spark/bin(spark版本2.4.5及以上)加入到PATH环境变量。
# This script provides a quick start guide for using the dcs jointcaller to perform joint calling on multiple gvcf files
# if you have any questions, please contact us at cloud@stomics.tech
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# dcs jointcaller is a wrapper of DPGT, which is a spark java application
# set JAVA_HOME and add spark/bin to PATH
export JAVA_HOME=/path/to/jdk8
export PATH=/path/to/spark/bin:$PATH
# Before using the dcs tool, you must obtain a license file and start the authentication proxy program
# on a node that can connect to the internet.
# Get the local IP address, which is used for license application.
# The output data field of the following command indicates the IP address.
# curl -X POST https://uat-cloud.stomics.tech/api/licensor/licenses/anon/echo?cliIp=true
# start the proxy program
# LICENSE=<path/to/license/file>
# DCS_HOST=0.0.0.0:8909
# $DCS_HOME/bin/licproxy --command start --license $LICENSE --bind $DCS_HOST --log-level INFO
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
# Set the following variables for dcs jointcaller
# gvcf directory
GVCF_DIR=$SCRIPT_DIR/data/gvcf
# target region
REGION=chr20:1000000-2000000
# reference fasta file
FA=$SCRIPT_DIR/data/ref/chr20.fa
# number of threads
nthreads=4
# java heap max memory
memory_size=8g
# Step 0: Set the working directory
workDir=$SCRIPT_DIR/result_jointcalling
mkdir -p $workDir
# Step 1: Set input file and output dir
# dcs jointcaller input file
GVCF_FOFN=$workDir/gvcf.fofn
ls ${GVCF_DIR}/*gz > ${GVCF_FOFN}
# dcs jointcaller output directory
OUT_DIR=$workDir/result
# Step 2: Run dcs jointcaller
$DCS_HOME/bin/dcs jointcaller \
--input ${GVCF_FOFN} \
--output ${OUT_DIR} \
--reference ${FA} \
--jobs ${nthreads} \
--memory ${memory_size} \
-l ${REGION}
4.5 SeqArc
参考脚本为quick_start_seqarc.sh,与WGS类似,修改其中的DCS_HOME和PROXY_HOST环境变量。
#!/bin/bash
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
nthreads=2
FQ1=$SCRIPT_DIR/data/fastq/simu.r1.fq.gz
FA=$SCRIPT_DIR/data/ref/chr20.fa
ARCFA=$SCRIPT_DIR/data/arcref/
mkdir -p $ARCFA
workDir=$SCRIPT_DIR/result_seqarc
mkdir -p $workDir
# Step 1: create index
$DCS_HOME/bin/dcs SeqArc index -r $FA -o $ARCFA
# Step 2: compress
$DCS_HOME/bin/dcs SeqArc compress -t $nthreads -r $ARCFA/chr20.fa -i $FQ1 -o $workDir/test
# step 3 : decompress
$DCS_HOME/bin/dcs SeqArc decompress -t $nthreads -r $ARCFA/chr20.fa -i $workDir/test.arc -o $workDir/simu.r1.fq
4.6 VarArc
#!/bin/bash
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
nthreads=4
VCF=$SCRIPT_DIR/data/vcf/demo.vcf.gz
workDir=$SCRIPT_DIR/result_vararc
mkdir -p $workDir
# Step 1: compress
$DCS_HOME/bin/dcs VarArc compress -t $nthreads -i $VCF -o $workDir/demo.arc --rowcnt 100 --blkcolcnt 2000
# step 2 : decompress
$DCS_HOME/bin/dcs VarArc decompress -t $nthreads -i $workDir/demo.arc -o $workDir/demo.vcf.gz
5. 发版说明
v2.0
支持多倍体物种的变异检测,体细胞变异检测部分开始支持多线程加速,aligner支持新的索引结构,增强稳定性。
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 功能开发 | aligner:支持低内存索引模式(运行速度略微降低),对资源要求更友好,并可以解决类似苏铁等基因组的bug variantCaller:支持多倍体物种的变异检测(参数:--ploidy)。添加WES数据的相应统计字段 report:支持输出WES数据的报告 util:bamStat功能(单独统计bam)与variantCaller保持一致,增加WES的统计字段 |
| 体细胞变异检测 | 功能开发 | 支持多线程加速模式,用--nthread参数设置,32线程下速度能达到原版本的8倍左右。 |
| 群体基因组联合分析 | 功能开发 | 支持多倍体物种(参数:--ploidy) |
| VarArc | 功能开发 | 新增该模块,支持对GVCF/VCF文件(文本或gz格式)的压缩和解压 |
备注:
1、WES数据的bam统计不要使用variantCaller的统计结果,要用util的bamStat功能单独统计
2、多倍体在variantCaller后,建议使用jointcaller来执行GVCF到VCF的操作
v1.5.0
支持使用加密狗(dongle)来做离线认证,达到单机使用本工具的目的。
| 模块 | 类型 | 描述 |
|---|---|---|
| 所有模块 | 功能开发 | 增加使用加密狗来进行离线license认证的功能,运行机器可以不联网使用DCS Tools |
| WGS/WES | 问题修复 | genotyper:修复内存占用较多的问题 |
v1.4.0
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 功能开发 | genotyper增加--include-non-variant-sites参数,与GATK的GenotypeGVCFs的同名参数功能类似,在VCF中保留GVCF的非变异block信息并将其展开,方便下游遗传病分析等场景。注意该参数开启时,需要设置ref文件(-R) |
| SeqArc | 问题修复 | 修复获取用户磁盘可用空间异常的bug |
v1.3.0
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 问题修复 | aligner: 1. 修复单行长度过长的fasta引起生成fai文件的问题。2. 修复arm架构欧拉操作系统的析构bug variantCaller: 1. 修复某些场景下的段错误。2. 修复某些物种情况下内存问题 |
| SeqArc | 功能开发 | 实现了windows解压工具 |
| SeqArc | 功能开发 | 支持arc直接解压为gz格式,并支持管道模式 |
| SeqArc | 功能开发 | 支持U碱基的压缩和解压 |
| 体细胞突变检测 | 功能开发 | 新增该模块 |
v1.2.0
支持arm指令架构,并支持样本量和数据量(压缩)统计,配合按样本量数据量的license模式。
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 问题修复 | bqsr:修复染色体长度超过2G时的bug variantCaller:修复-L参数部分场景下的bug,修复gvcf索引文件问题 |
| 功能开发 | aligner:增加共享内存功能,需要开启--useSharedMemIndex参数 variantCaller:1.增加15x覆盖度统计结果 2.兼容有overlap的bed文件 report:更新报告版本号,增加15x覆盖度统计 | |
| SeqArc | 功能开发 | 实现了单独的解压工具 |
| 功能开发 | 实现了基因库的统计功能 | |
| JointCalling | 问题修复 | 修复了分区end bug |
v1.1.0
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 问题修复 | 解决aligner内存泄漏问题 |
| 问题修复 | 解决aligner在构建某些ref fasta索引时出现卡死问题 | |
| SeqArc | 功能开发 | 使用simde来实现跨平台指令集 |
| 功能开发 | 实现了单独的解压工具 | |
| 功能开发 | 实现了基因库的统计功能 |
v1.0.1
改动模块:aligner, report
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 问题修复 | 修复了aligner某种场景下coredump的问题 |
| 问题修复 | 修复了aligner输出文件为不带路径的纯文件名时的问题 | |
| 问题修复 | 将aligner生成的bam索引文件重命名为*.bam.bai,不再是*.bai | |
| 问题修复 | 修复report模块深度累积频率图潜在问题 | |
| 问题修复 | quick_start包中wgs脚本中增加set -e,遇到错误时及时退出 |
v1.0.0
| 模块 | 类型 | 描述 |
|---|---|---|
| WGS/WES | 功能开发 | 新增比对和变异相关信息统计功能 |
| SeqArc | 功能开发 | 按照avs-g标准,重构了项目框架 |
| 功能开发 | 增加了长读长数据的压缩功能 | |
| 功能开发 | 调整了参数解析方式,增加了子命令和长短参数 |
6. 注意事项
6.1 WGS/WES
- 比对时需要参考序列索引文件,用aligner提前准备好,避免找不到文件报错
- bqsr步骤默认只输出recal.table,如果需要碱基质量校正后的bam文件,需要设置--out-bam参数
- 在WES场景下做比对结果统计时,可以有两种方式,一是在variantCaller步骤指定interval参数为wes bed文件,二是单独运行bam-stat工具,设置--bed参数。如果不添加bed文件,默认统计的是全基因组范围,导致覆盖度深度等信息结果不准
6.2 Joint Calling
- 输入的gvcf文件需要带上tbi索引文件。
- 参考基因组fasta需要fai和dict文件。
- 样本量很大时,建议参考7中的运行示例设置内存,避免内存设置过小导致内存溢出。
6.3 SeqArc
- 压缩输入的fastq文件,文件名后缀需要是 *.fq *.fastq *.fq.gz *.fastq.gz,否则认为文件类型不正确
- 输入的参考序列文件,需要是文本文件,文件名后缀需要是*.fa *.fasta,否则认为文件类型不正确
- 解压输入的arc文件,需要是本程序压缩的结果文件,否则认为文件类型不正确。
- 解压输入的参考序列文件,需要和压缩的参考文件保持一致,如果不一致,会报错提示:[ref load md5 error]
- fastq如果不完整,会报错提示[read fastq error]
- read如果seq和qual不等长,会报错提示[seqlen isnot equal quallen]
- 压缩,解压如果文件读写异常,会报错提示[read file err] [write file err]
- 解压时,如果数据块校验失败,会提示报错[block check fail],这可能是压缩时异常,或者压缩数据没有正确保存。
- 需要设置 ulimit -n, 对于运行时的崩溃,会生成对应的coredump。
6.4 License Proxy
工具 DCS Tools 或 lickit 无法连接 licproxy。
如果您遇到了类似如下错误提示:
- connection refused
- couldn't connect to server
请按照如下步骤进行排查:
- 在运行 DCS Tools 或 lickit 的机器通过如下命令行检查与 License 代理是否联通
curl -v http://$PROXY_HOST/api/licensor/licenses/anon/pool
请将 $PROXY_HOST 替换成您部署 licproxy 的内网 IP 地址和端口,例如 172.20.9.149:8909,而不是您在获取 License 文件前执行获取联网机器的出口 IP 地址!
如果运行 DCS Tools 或 lickit 的机器和运行 licproxy 在同一台机器,您可以将 $PROXY_HOST 替换为 127.0.0.1:8909
预期返回包含 Success,若提示无法连接,则请继续按下面的步骤检查 License 代理进程是否存在。
- 在部署 licproxy 的机器上查看进程是否存在
ps aux | grep licproxy
若进程不存在,则请先按照上文 1.4 软件部署进行操作。若进程存在,则请联系您们的 IT 运维确保运行DCS Tools 或 lickit 的机器可以通过上文步骤 1 访问 licproxy 所在机器,若网络不通则请向 IT 运维申请开通。
- 检查投递任务的脚本或命令行是否正确设置变量 PROXY_HOST
请检查您使用的脚本或命令行是否覆盖环境变量 PROXY_HOST,例如 licproxy 部署在 172.20.9.149:8909,那么您在运行脚本或命令行之前务必先设置:
export PROXY_HOST=172.20.9.149:8909
./path/to/your
# 或者
PROXY_HOST=172.20.9.149:8909 /path/to/your
License 认证常见错误提示
Permission denied(无权限或验证失败)
Size must be greater than 0(提交线程数必须大于零)
Pool size exceeded(提交线程数大于当前线程数)
License expired(许可证已过期)
Unknown error(未知错误)
7. 性能和运行时间
7.1 WGS
在阿里云的ecs.i4g.8xlarge(32核,128G内存)机型上,存储使用SSD,分别对HG001 30X WGS NovaSeq、HG001 30X DNBSEQ-G400和HG005 40X WGS数据进行测试,dcs tools需要1.82小时、1.91小时和2.45小时完成从FASTQ到VCF的过程。
| 数据 | 运行时间(h) | 线程数 | 最大内存 |
|---|---|---|---|
| HG001 30X NovaSeq | 1.82 | 32 | 101 |
| HG001 30X DNBSEQ-G400 | 1.91 | 32 | 100 |
| HG005 40X DNBSEQ-G400 | 2.45 | 32 | 120 |
针对低内存索引aligner版本,在Intel(R) Xeon(R) Gold 6442Y机型上,对HG001 30X WGS NovaSeq数据进行测试。
| 工具 | 运行时间(h) | 线程数 | 最大内存(GB) |
|---|---|---|---|
| aligner | 0.83 | 32 | 97.99 |
| aligner(low memory index) | 1.11 | 32 | 39.22 |
7.2 SeqArc
在阿里云的ecs.c6a.xlarge(4核,8G内存)机型上,针对NA12878 (30X WGS)数据进行压缩和解压测试:
压缩测试结果:
| 输入数据 | 时长 | 压缩率(相比.gz输入数据) |
|---|---|---|
| r1.fq.gz(27G) | 27m 24s | 5.17 |
| r2.fq.gz (30G) | 29m 14s | 4.10 |
解压测试结果:
| 输入数据 | 时长 |
|---|---|
| r1.fq.arc | 14m 32s |
| r2.fq.arc | 14m 25s |
7.3 Joint Calling
在公开数据1KGP样本集和内部的多个大规模人群样本集上测试,dcs jointcaller(DPGT)的计算性能表现如下表:
| 数据 | 样本数 | 进程数(每个进程分配6g内存) | 核时 | 运行时间 |
|---|---|---|---|---|
| 1KGP | 2504 | 256 | 6963 | 27.2 hours |
| Internal 10K | 9165 | 256 | 21376 | 83.5 hours |
| Internal 52K | 52000 | 4400 | 407760 | ~6 days |
| Internal 105K | 105000 | 3675 | 1026155 | ~19 days |
7.4 VarArc
在本地linux集群,对群体变异VCF数据(来自CKB项目)测试,VarArc的表现如下表:
| 名称 | 数据来源 | gz数据大小/GB | 染色体 | 样本数 | 位点数 | 字段特征 | 工具 | comp_size(GB) | 压缩比 | comp_RealTime(s) | comp_CPUTime(s) | comp_CPUUsage | comp_MaxRAM(GB) | 压缩速度 (以gz为分子) MB/s | decomp_RealTime(s) | decomp_CPUTime(s) | decomp_CPUUsage | decomp_MaxRAM(GB) | 解压速度 (以gz为分子) MB/s |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| data1 | CKB | 15.00 | chr1 | 6w | 8w | 变异含GT,AD,DP等字段 | VarArc | 6.32 | 2.37 | 1251 | 31221 | 2510% | 29.2 | 12.3 | 352 | 5091 | 1466% | 7.4 | 43.7 |
| bcftools | 18.80 | 0.80 | 510 | 4630 | 916% | 0.2 | 30.1 | 357 | 3745 | 1058% | 0.1 | 43.0 | |||||||
| data2 | CKB | 25.90 | chr10 | 10.5w | 8w | 变异含GT,AD,DP等字段 | VarArc | 11.17 | 2.32 | 1783 | 48871 | 2754% | 94.39 | 14.9 | 587.31 | 8533.3 | 1468% | 9.5 | 45.2 |
| bcftools | 32.33 | 0.80 | 832 | 7933 | 960% | 0.16 | 31.9 | 606.16 | 6713.36 | 1116% | 0.1 | 43.8 | |||||||
| data3 | CKB | 60.70 | chr22 | 10.5w | 524w | 变异仅含GT字段 | VarArc | 53.79 | 1.13 | 13423 | 372176 | 2778% | 31.03 | 4.6 | 3881.66 | 108601.14 | 2815% | 3.2 | 16.0 |
| bcftools | 52.40 | 1.16 | 10189 | 39974 | 397% | 0.06 | 6.1 | 8186.85 | 43343.32 | 535% | 0.1 | 7.6 |
在本地linux集群,对单样本gVCF数据(来自CKB项目)测试,VarArc的表现如下表:
| gz数据大小/GB | 工具 | comp_size(GB) | 压缩比 | comp_RealTime(s) | comp_CPUTime(s) | comp_CPUUsage | comp_MaxRAM(GB) | 压缩速度 (以gz为分子) MB/s | decomp_RealTime(s) | decomp_CPUTime(s) | decomp_CPUUsage | decomp_MaxRAM(GB) | 解压速度 (以gz为分子) MB/s |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 51.6 | VarArc | 15.52 | 3.3 | 4141.1 | 112007.3 | 2690% | 14.1 | 12.8 | 1213.4 | 10153.0 | 831% | 8.9 | 43.6 |
| bcftools | 54.25 | 1.0 | 2438.3 | 10293.2 | 436% | 0.0 | 21.7 | 1474.8 | 7947.5 | 570% | 0.0 | 35.8 |
7.5 Mutect2
WES数据(normal和tumor各约11G bases)
| 版本 | 运行时间 |
|---|---|
| gatk-Mutect2 | 5824 s |
| dcs-Mutect2 (32线程) | 726 s |
WGS数据(normal:221G bases,tumor:642G bases)
| 版本 | 运行时间 |
|---|---|
| gatk-Mutect2 | 57.3 h |
| dcs-Mutect2 (32线程) | 7.5 h |
8. FAQ
8.1 部署相关问题
1. DCS Tools对平台的要求?
DCS Tools 设计为运行在Linux系统的可执行程序;
推荐的Linux发行版为:Centos 7,Debian 8,Ubuntu 14.04及以上的版本。
2.对系统硬件的要求?
cpu 至少有8核,mem至少有128G,并且建议使用读写速度更快的SSD硬盘。
3.软件部署环节是否需要联网?
DCS Tools运行时会进行 License 验证,以确保合法性,因此需要一台可以联网(用来与验证服务端通信)的机器,其它机器(往往是计算节点)不需要联网,保证能与联网机器通信即可。当然,单台联网机器也可以部署本软件。
4.软件部署都需要哪些文件?
软件包+License 文件,可以通过云平台(cloud@stomics.tech)或者销售经理获取。
我们支持30天免费试用,可联系销售经理。
5.license proxy部署成功后,为什么运行DCS Tools时还会出现license相关错误?
在license proxy部署后,可以通过文档中提供的命令,确认与云平台的连接是否成功。另外每个license都有对应线程数上限,如果使用达到了上限,也会导致新任务无法成功启动,需要等待正在运行的任务结束或者扩充线程数。
8.2 工具相关问题
1.Joint calling输出的不是合并的gvcf,而是vcf?
由于传统一次性生成多样本 gvcf 文件计算量大且占用存储空间,我们未采用该策略来获取所有样本的 gvcf 结果。建议用户保留各样本的 gvcf 文件,后续若需对特定样本进行联合调用(joint calling),直接基于单样本 gvcf 文件运行即可。
2.WGS是否支持非二倍体物种的分析?
DCS Tools的变异检测流程是基于二倍体检测模型构建的,因此WGS暂无法对多倍体样本开展有效的变异检测工作。
3.WGS数据运行时间为啥比文档中的要明显长?
除数据量因素外,WGS流程的运行时间还会受到存储设备读写速度的制约。在我们的测试环境中,采用了具备高速读写性能的SSD固态硬盘,以尽可能降低该因素对流程运行时间的影响。
4.压缩测试用的是什么平台的数据?
目前DCS Tools所用压缩方法已实际压缩数据达几十PB,在开发阶段开展的大规模测试中,最具代表性的数据集为 AVS-G 参考测试集。该数据集抽取自 NCBI-SRA 库,涵盖了13个最具代表性的物种,包含39组 FASTQ 数据。在样本属性、实验方法、测序技术、测序代数以及测序平台等方面,集尽可能全面地覆盖了 NCBI 上的各类情况。
作为参考,DCS Tools压缩方法在该测试集上的压缩率为8.11,而 gzip 的压缩率为4.15。鉴于物种、测序平台等不同属性均会对压缩率产生一定影响,建议用户结合DCS Tools及同类工具在自身数据上的实际测试结果进行决策。
AVS-G 参考测试集相关详细资料可通过销售经理,或者公众号后台联系我们获取。
5.压缩100GB FASTQ 源文件,DCS Tools与常用的 pigz 相比如何?
在32线程环境对人类100GB WGS的PE数据进行测试,测试结果如下:
| 测试数据 | pigz压缩率 | pigz压缩速度 | pigz CPU占用率 | DCS Tools压缩率 | DCS Tools压缩速度 | DCS Tools CPU占用率 |
|---|---|---|---|---|---|---|
| BGI-SEQ500(ERR2755197) | 3.93 | 450MB/s | 3224% | 5.66 | 937MB/s | 3177% |
| NovaSeq(SRR10965088) | 4.9 | 220MB/s | 3175% | 21.15 | 794MB/s | 3147% |
6.同平台的不同FASTQ文件压缩率会不一样么?主要原因有哪些?
FASTQ文件主要包含测序序列和质量分数两个部分,差异也出自这里。
首先是测序序列,DCS Tools推荐使用在压缩&解压时加入参考序列的模式,可以有效提升序列的压缩率,而参考序列是该物种的基因组序列,那么对于转录组测序来说这个模式的效果就比较差,而宏基因组测序难以使用该模式。即便对于基因组测序来说,不同平台/型号的测序准确率也是不同的,一般来说测序越准确压缩率就越好。
然后是质量分数,首先不同型号的质量分数的种类是不同的,质量值种类一般分为40和4两种,显然种类越少压缩率越好,这也是它们压缩率差异的主要来源。另外,质量分数本身随机性较强,这也与测序仪强相关,随机性越强的数据压缩率就越差。