SAW_spatial_scRNA_V1
芯片单细胞流程使用手册(V1.1.0)
SAW_spatial_scRNA_V1
第一章 产品信息
产品描述
Stereo-cell技术是基于DNB芯片成功研发组织空间原位捕获技术Stereo-seq,围绕自主底层技术DNB芯片的创新性研发,使应用方向拓展至单细胞组学生物大分子的捕获,匹配不同通量,匹配大尺寸细胞,高细胞回收率,该技术优势:
•不依赖特定细胞分离包裹设备,利用包被多聚赖氨酸芯片表面进行细胞均质分散和捕获。
•在实验流程中整合细胞染色和成像。
实现细胞组学技术在早诊、慢性疾病筛查等方面的液体活检新应用。
本流程是基于 SAW-ST-V7 转录组分析流程+圈细胞分析流程构建的芯片单细胞分析流程,采用 下机fastq 数据作为输入的全自动化流程,搭建的芯片单细胞分析流程,流程特征如下:
1、只支持umi在read1的使用场景;
2、支持对reads进行rRNAremove;
3、适配ImageQC 3.0.0及以上版本的图像,图像类型包括(ssDNA、DAPI、mIF、H&E);暂时不支持依图像圈细胞功能。
4、适配全物种的圈细胞分析;
5、支持对细胞进行注释并生成分析报告。
若您将SAW V7流程和圈细胞流程分开运行,官方同时提供三个配套的圈细胞流程,可按需选择使用,分为三个子流程:
以SAWv7流程的结果文件中SN.tissue.gem.gz作为输入进行圈细胞处理最终生成报告。
6、Chips_scRNA_singlespecies_unlimited_S1:适配非人鼠物种的圈细胞流程。
7、Chips_scRNA_singlespecies:适配人鼠物种的圈细胞流程。
8、Chips_scRNA_cellline:适配人鼠混合细胞系的圈细胞流程。
注意事项
当前流程仅支持SAWv7流程的全自动芯片单细胞流程,适配芯片细胞组学试剂盒产出数据的分析。
当前流程最新版本为:V1.1.0。
主流程名称:SAW_spatial_scRNA_V1
子流程名称:Chips_scRNA_singlespecies_unlimited_S1、Chips_scRNA_singlespecies和Chips_scRNA_cellline
Q: 申请要建ref,可以参考哪个资料说明?
A: 可以参考这个sop文档 https://alidocs.dingtalk.com/i/nodes/AR4GpnMqJzM3oe71CONRd01nVKe0xjE3?utm_scene=team_space
第二章 产品介绍
芯片单细胞原理
Stereo-cell是一种基于高密度时空芯片的单细胞(核)3’转录组测序技术。
该技术通过创新性三步流程实现单细胞解析:
- 空间编码:基于微米级精密工艺,在芯片表面构建携带唯一坐标标识(CID)的Poly(dT)探针阵列,形成可溯源的时空坐标系统(即时空芯片);
- 原位捕获:固定于芯片的单细胞(核)经透化处理后,释放的mRNA被对应空间坐标的探针原位捕获,通过逆转录生成含CID和分子标签(UMI)的cDNA文库;
- 智能解析:结合MASK文件坐标映射及UMI校正扩增偏差,运用深度学习驱动的图像分割算法精确定义细胞边界,最终构建单细胞表达矩阵。
芯片单细胞应用场景
Stereo-cell不仅适用于传统的单细胞测序场景,还在以下场景拥有核心技术优势:
- 零设备依赖:突破传统单细胞技术对细胞尺寸及形态的限制,直接兼容细胞(核)及复杂形态样本;
- 动态通量设计:单芯片(1cm²)支持200-15,000细胞级柔性检测,适配从稀有样本到高通量研究场景;
- 多组学融合:通过细胞核/膜荧光染色与转录组协同分析,实现基因表达与细胞形态的跨模态关联。
流程原理
Stereo-cell的流程,是基于时空芯片流程得到的表达矩阵,利用RNA表达的聚集程度做细胞分割。目前Stereo-cell流程(SAW_Stereo-cell_V1)是将转录组流程(SAW-ST-V7)以及研发单细胞流程(Chips_scRNA_singlespecies_unlimited)整合得到,使用户直接输入SAW的入参(如:芯片位置mask文件,fastq文件,图像ipr以及tar.gz文件等参数),得到:
- RNA的表达矩阵;
- 转录组基本质控的报告;
- 单细胞的细胞分割矩阵;
- 单细胞质控以及单细胞聚类报告。
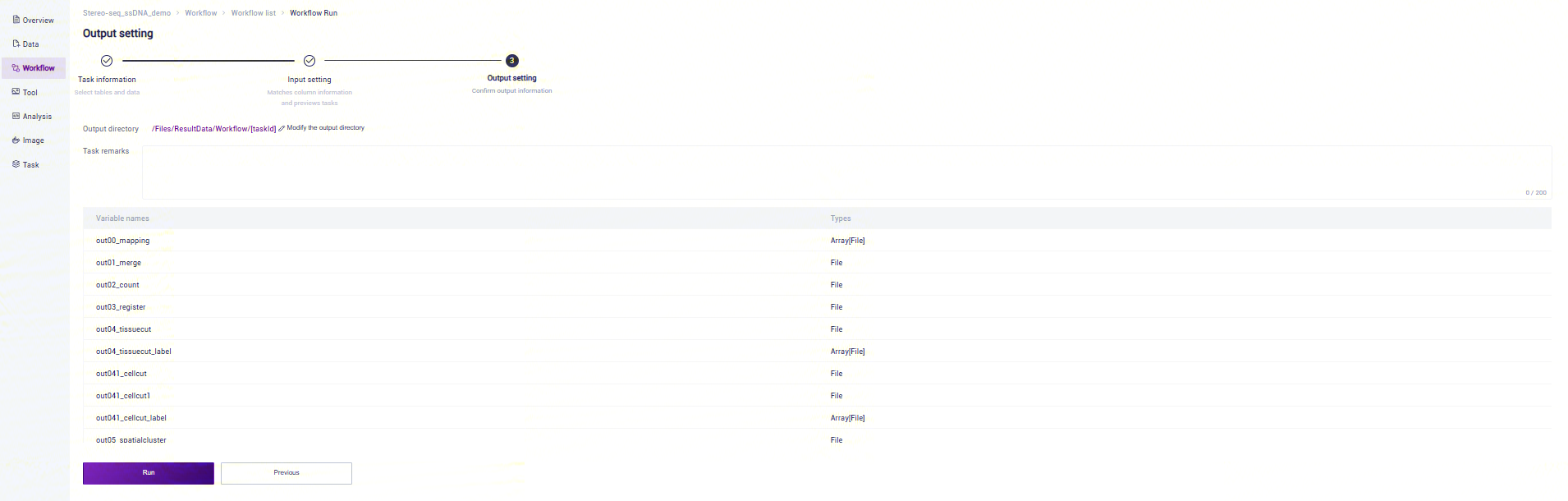
流程产出结果
目录00.*-07.*为转录组数据处理输出目录,同SAW-ST-V7产出的结果内容。
目录08.scRNA_result为Stereo-cell输出的结果文件,以下为目录结构和说明:
report
- *.cellsegment.png # RNA圈细胞算法得到的细胞边界与RNA表达的merge图,可以查看RNA聚集程度以及圈细胞效果。
- *.html # 芯片单细胞质控报告。
RNA_Segment
- *_mask_corr10.tif #细胞修正10个pixel的mask。
- *_mask_corr20.tif #细胞修正20个pixel的mask。
- *_mask_corr30.tif #细胞修正30个pixel的mask。
- *_mask.tif # RNA圈细胞算法得到的原始细胞mask。
- cell_area_corr10.txt #细胞修正10个pixel的细胞DNB数目统计。
- cell_area_corr20.txt #细胞修正20个pixel的细胞DNB数目统计。
- cell_area_corr30.txt #细胞修正30个pixel的细胞DNB数目统计。
- cell_area.txt # RNA圈细胞算法得到的原始细胞DNB数目统计。
- Cell_GetExp_gene_with_background_corr10.txt # 细胞gem矩阵,修正10个pixel。
- Cell_GetExp_gene_with_background_corr20.txt# 细胞gem矩阵,修正20个pixel。
- Cell_GetExp_gene_with_background_corr30.txt# 细胞gem矩阵,修正30个pixel。
- Cell_GetExp_gene_with_background.txt# 细胞gem矩阵,原始未修正。
第三章 使用说明书
SAW_spatial_scRNA_V1分析流程通过DCS云平台系统进行样本数据输入及报告输出的全流程管理。下面具体介绍基于DCS云平台系统使用SAW_spatial_scRNA_V1主流程及配套圈细胞子流程的操作指南。
指南概述
概述
本章介绍如何使用SAW_spatial_scRNA_V1分析流程进行分析。在使用之前,请认真阅读并理解其中内容,保证能够正确使用SAW_spatial_scRNA_V1分析流程。
使用场景一:手动投递
操作共包括四个步骤:上传数据、样本信息录入、启动分析。完成样本信息录入后,运行任务,当客户在看到任务状态为completed 时,代表任务已完成,即可查看可视化及报告部分(详见2.4部分)。
步骤一:上传数据
- 点击导航栏**【Data】,进入数据管理页面,进入目标文件夹,点击右上角【+Add files】-【Tool upload】**上传数据(图3-1):
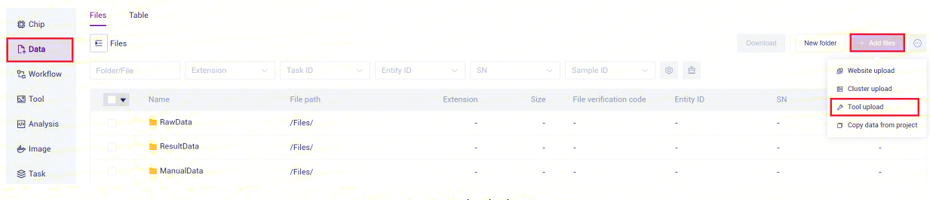
- 点击**【Upload】浏览并选择所需文件(图3-2),上传完成后文件会在目标文件夹中显示(如首次上传,则需点击【Install and start transport client】**安装所需工具):
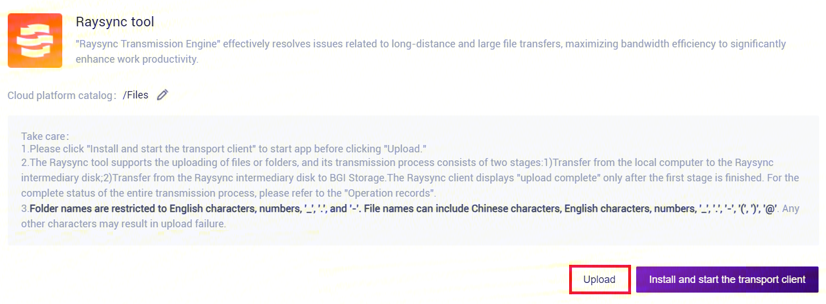
SAW_spatial_scRNA_V1流程运行:
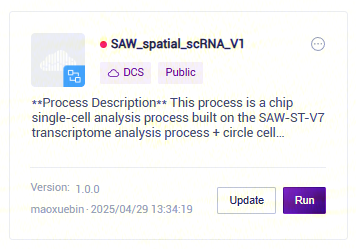
【1】SAW_spatial_scRNA_V1样本信息录入
- 点击导航栏**【Workflow】**进入流程分析页面,在搜索框输入 SAW_spatial_scRNA_V1,点击【Run】**(图3-3):
- 选择Run workflow,输入Entity ID,点击**【Next】**(图3-4):
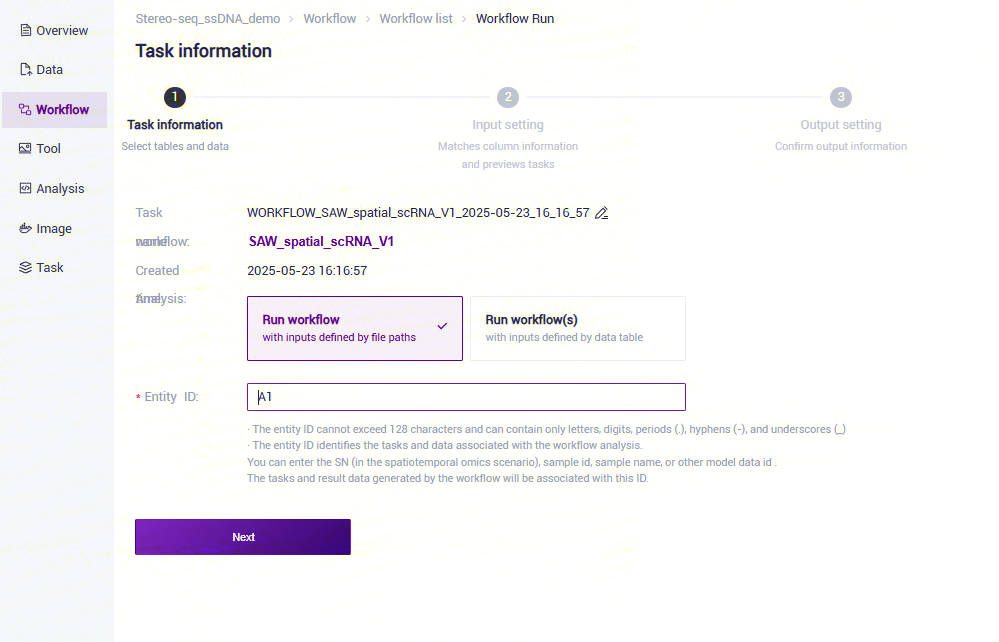
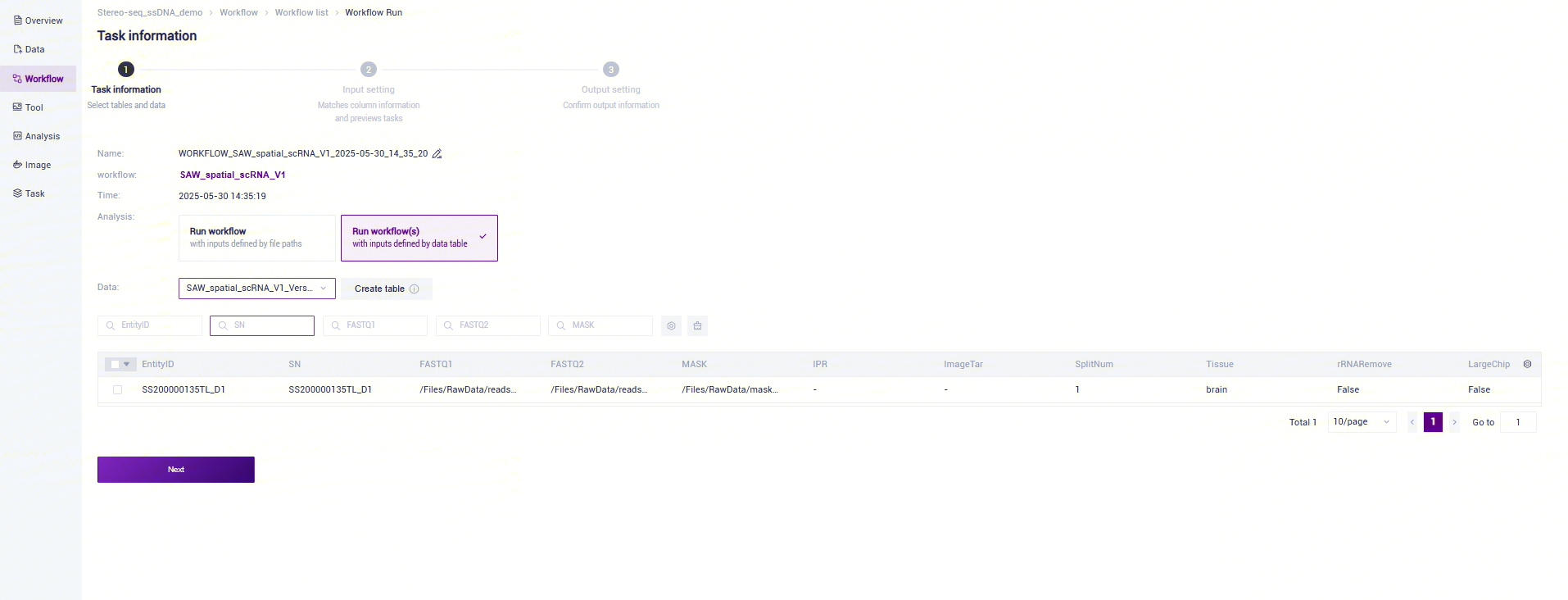
- 录入样本信息,完成后点击**【Next】**(图3-5):
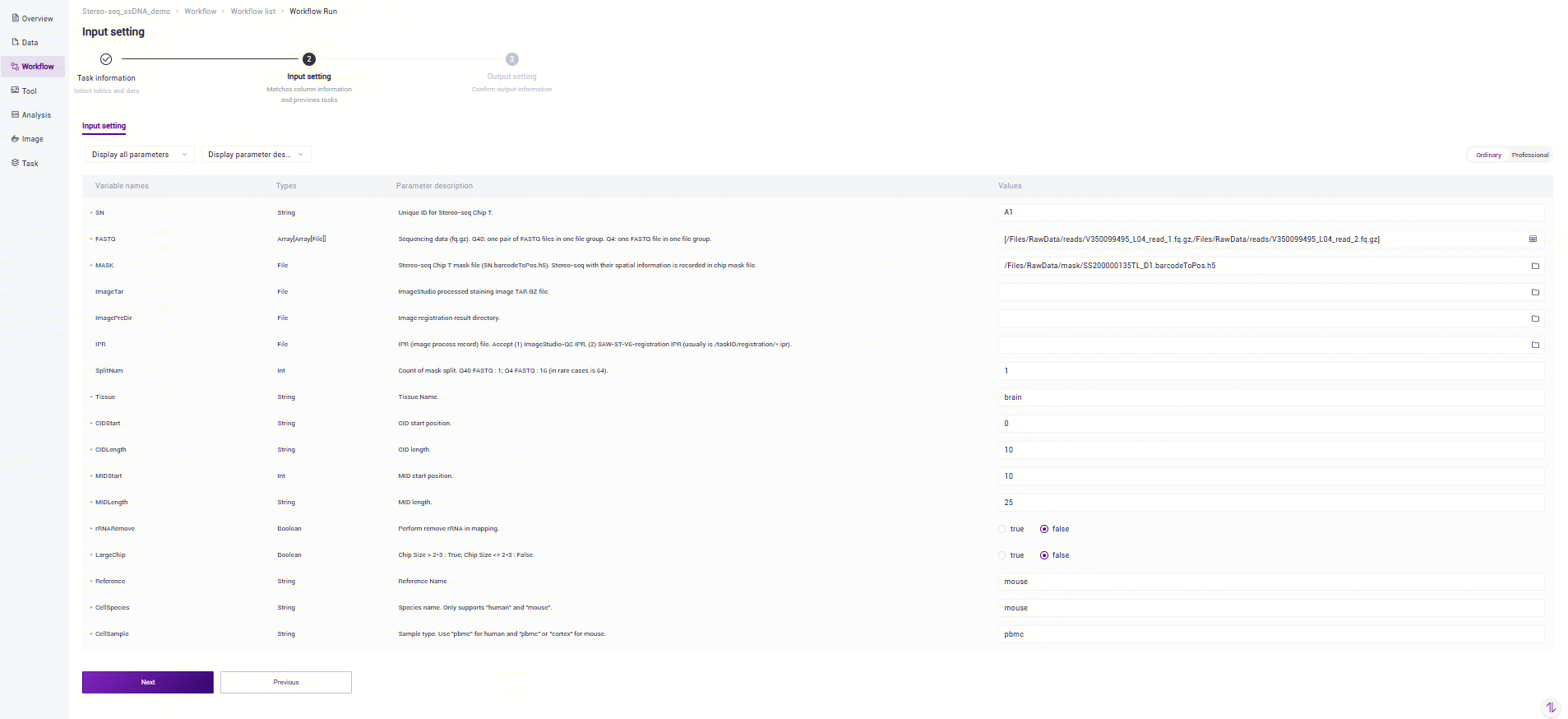
信息录入参数说明:
- SN:Unique ID for Stereo-seq Chip T.;
- FASTQ :Sequencing data (fq.gz). Q40: one pair of FASTQ files in one file group. Q4: one FASTQ file in one file group;
- MASK:Stereo-seq Chip T mask file (SN.barcodeToPos.h5). Stereo-seq with their spatial information is recorded in chip mask file.
- ImageTar:ImageStudio processed staining image TAR.GZ file.
- IPR:IPR (image process record) file. ImageStudio-QC IPR.
- SplitNum:Count of mask split. Q40 FASTQ : 1; Q4 FASTQ : 16 (in rare cases is 64).
- Tissue:Tissue Name.
- rRNARemove:Perform remove rRNA in mapping.
- LargeChip:Chip Size > 2*3 : True; Chip Size <= 2*3 : False.
- Reference:Reference Name.
- CellSpecies:Species name. Only supports "human" and "mouse".
- CellSample:Sample type. Use "pbmc" for human and "pbmc" or "cortex" for mouse.
- ScMem:Memory usage of single cell analysis.
【2】启动分析
点击**【Run】**启动分析(图3-6):
Chips_scRNA_singlespecies_unlimited_S1流程运行:
【1】Chips_scRNA_singlespecies_unlimited_S1样本信息录入
- 点击导航栏**【Workflow】**进入流程分析页面,在搜索框输入Chips_scRNA_singlespecies_unlimited_S1 ,点击【Run】(图3-7):
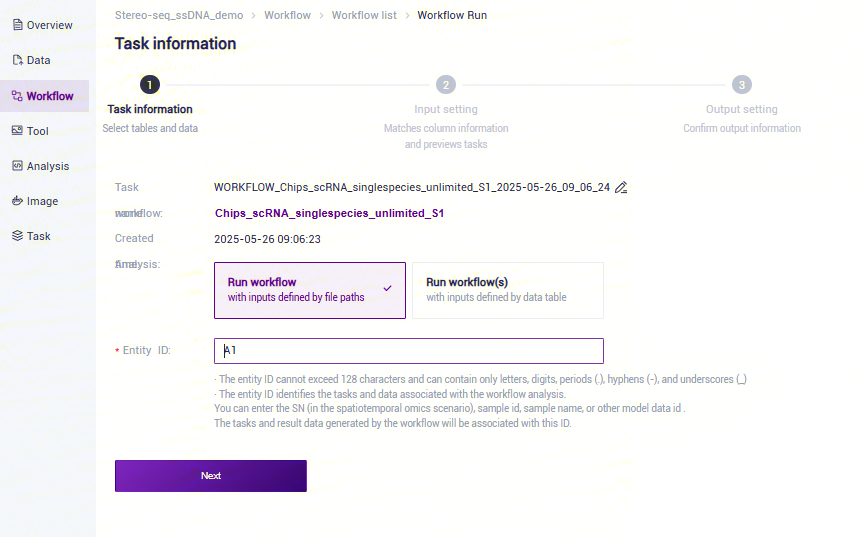
- 选择Run workflow,输入Entity ID,点击**【Next】**(图3-8):
- 录入样本信息,完成后点击**【Next】**(图3-9):
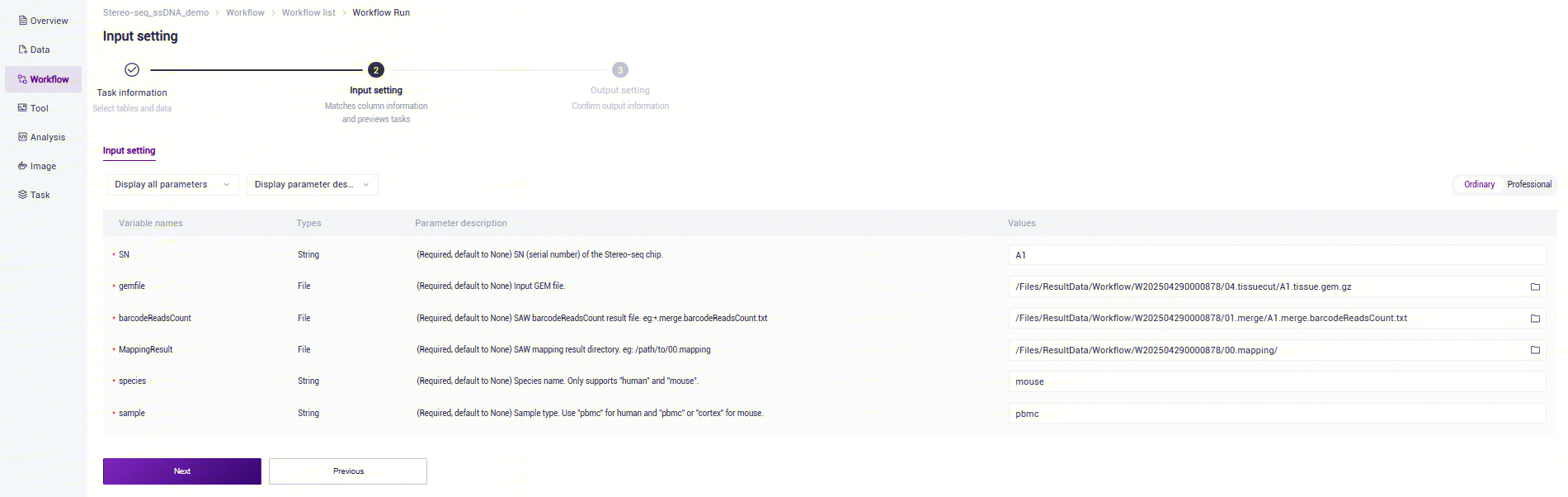
信息录入参数说明:
- SN:Unique ID for Stereo-seq Chip T.;
- gemfile :(Required, default to None) Input GEM file.
- barcodeReadsCount:(Required, default to None) SAW barcodeReadsCount result file. eg:*.merge.barcodeReadsCount.txt.
- MappingResult:(Required, default to None) SAW mapping result directory. eg: /path/to/00.mapping.
- species:(Required, default to None) Species name. Only supports "human" and "mouse".
- sample:(Required, default to None) Sample type. Use "pbmc" for human and "pbmc" or "cortex" for mouse.
【2】启动分析
点击**【Run】**启动分析(图3-10):
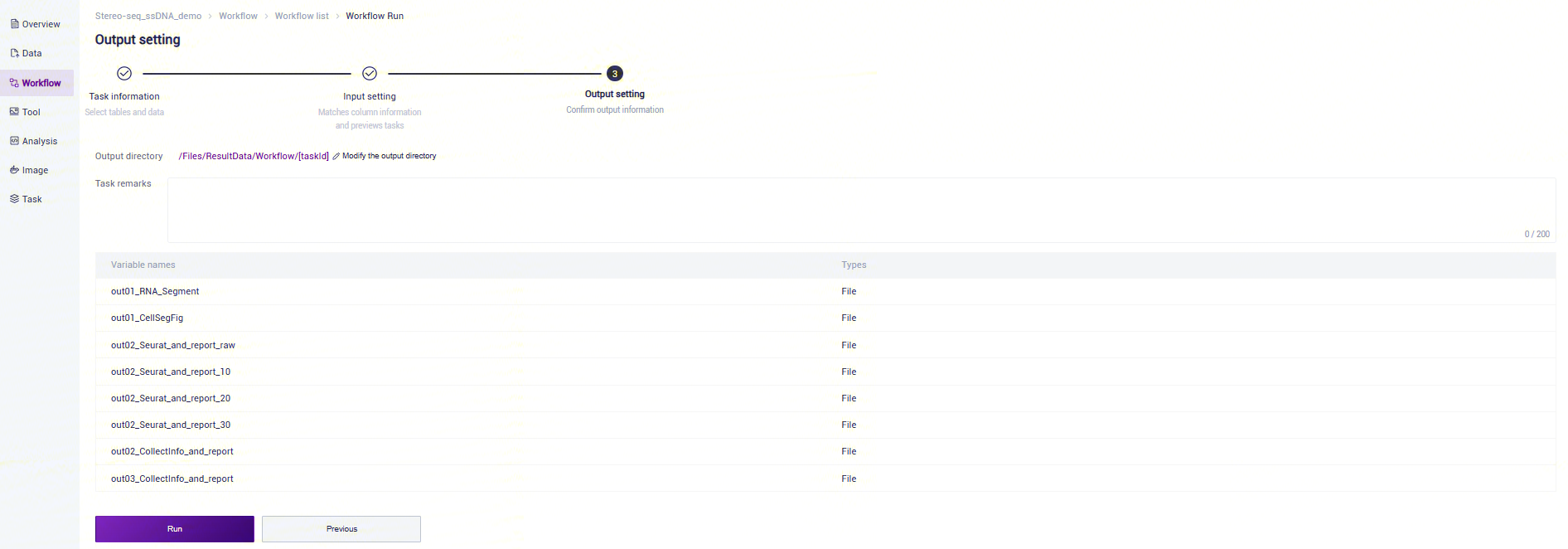
Chips_scRNA_singlespecies流程运行:
【1】Chips_scRNA_singlespecies样本信息录入
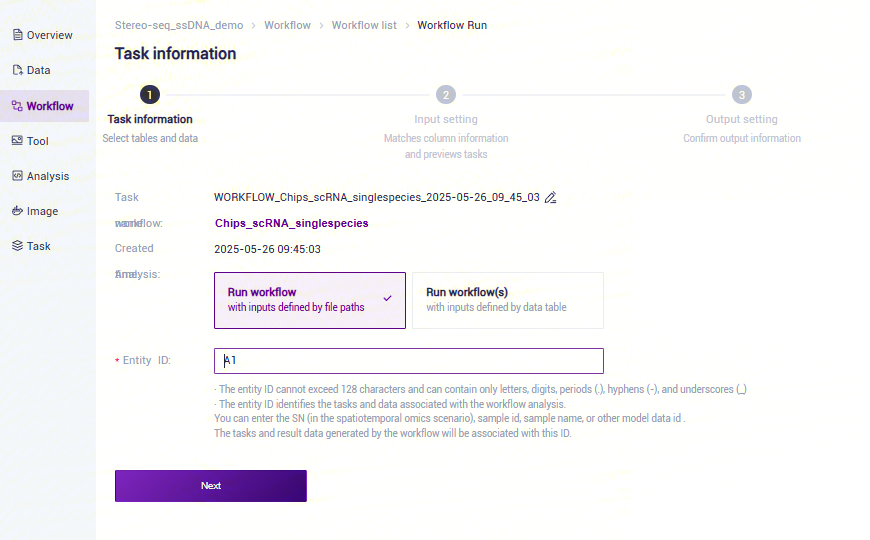
- 点击导航栏**【Workflow】**进入流程分析页面,在搜索框输入 Chips_scRNA_singlespecies,点击【Run】**(图3-11):
- 选择Run workflow,输入Entity ID,点击**【Next】**(图3-12):
- 录入样本信息,完成后点击**【Next】**(图3-13):
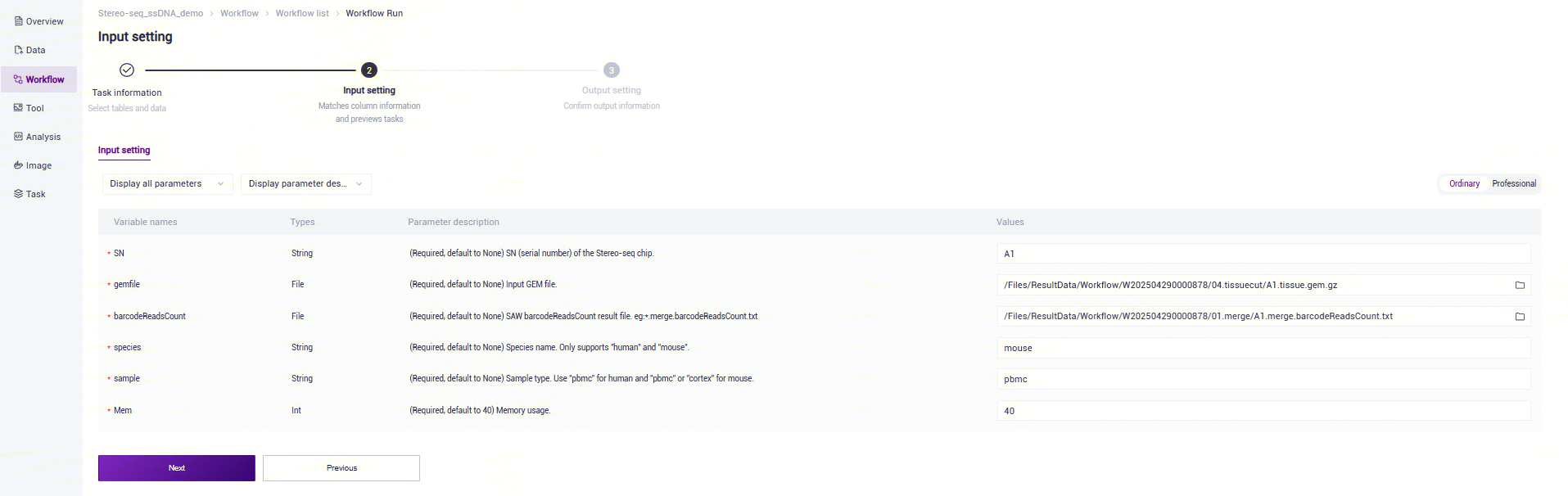
信息录入参数说明:
- SN:Unique ID for Stereo-seq Chip T.;
- gemfile :(Required, default to None) Input GEM file.
- barcodeReadsCount:(Required, default to None) SAW barcodeReadsCount result file. eg:*.merge.barcodeReadsCount.txt.
- MappingResult:(Required, default to None) SAW mapping result directory. eg: /path/to/00.mapping.
- species:(Required, default to None) Species name. Only supports "human" and "mouse".
- sample:(Required, default to None) Sample type. Use "pbmc" for human and "pbmc" or "cortex" for mouse.
- Mem:(Required, default to 40) Memory usage.
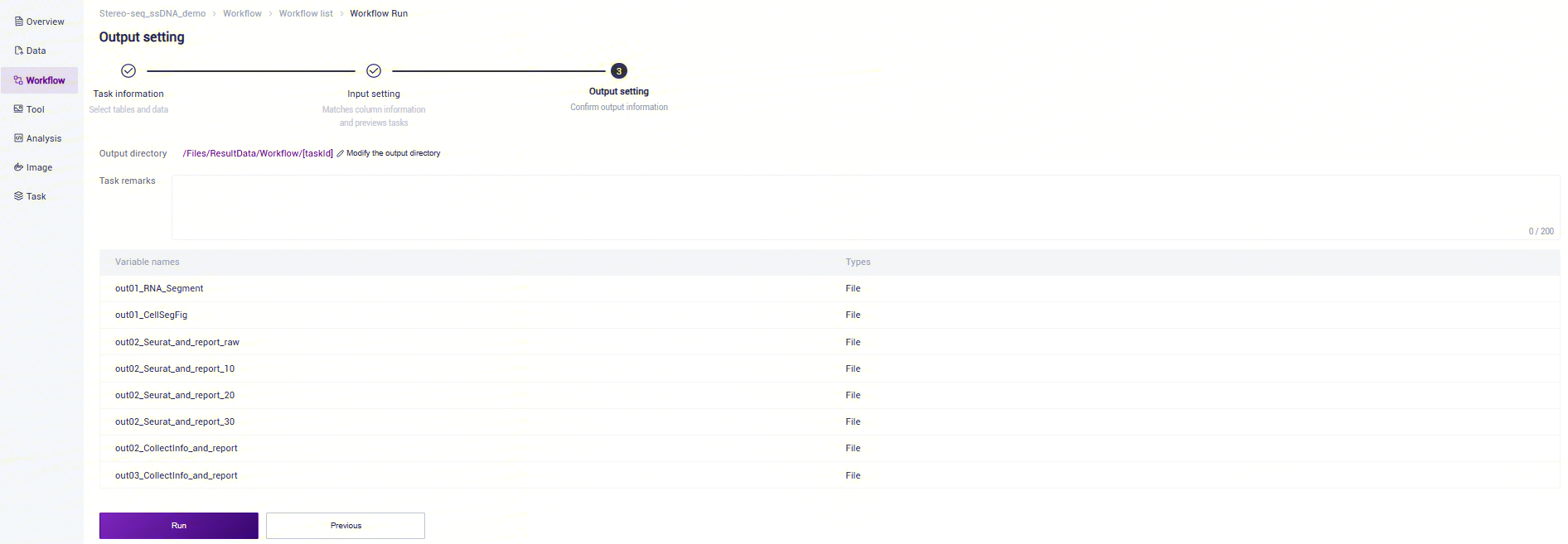
【2】启动分析
点击**【Run】**启动分析(图3-14):
使用场景二:表格投递
操作共包括五个步骤:上传数据、下载样本模板、填写并导入样本模板、启动分析。完成样本模板导入后,可批量运行任务,当客户在看到任务状态为completed 时,代表任务已完成,即可查看报告和可视化部分。
步骤一:上传数据
与使用场景一(手动投递)步骤一一致(详见3.2.1步骤一:上传数据)。
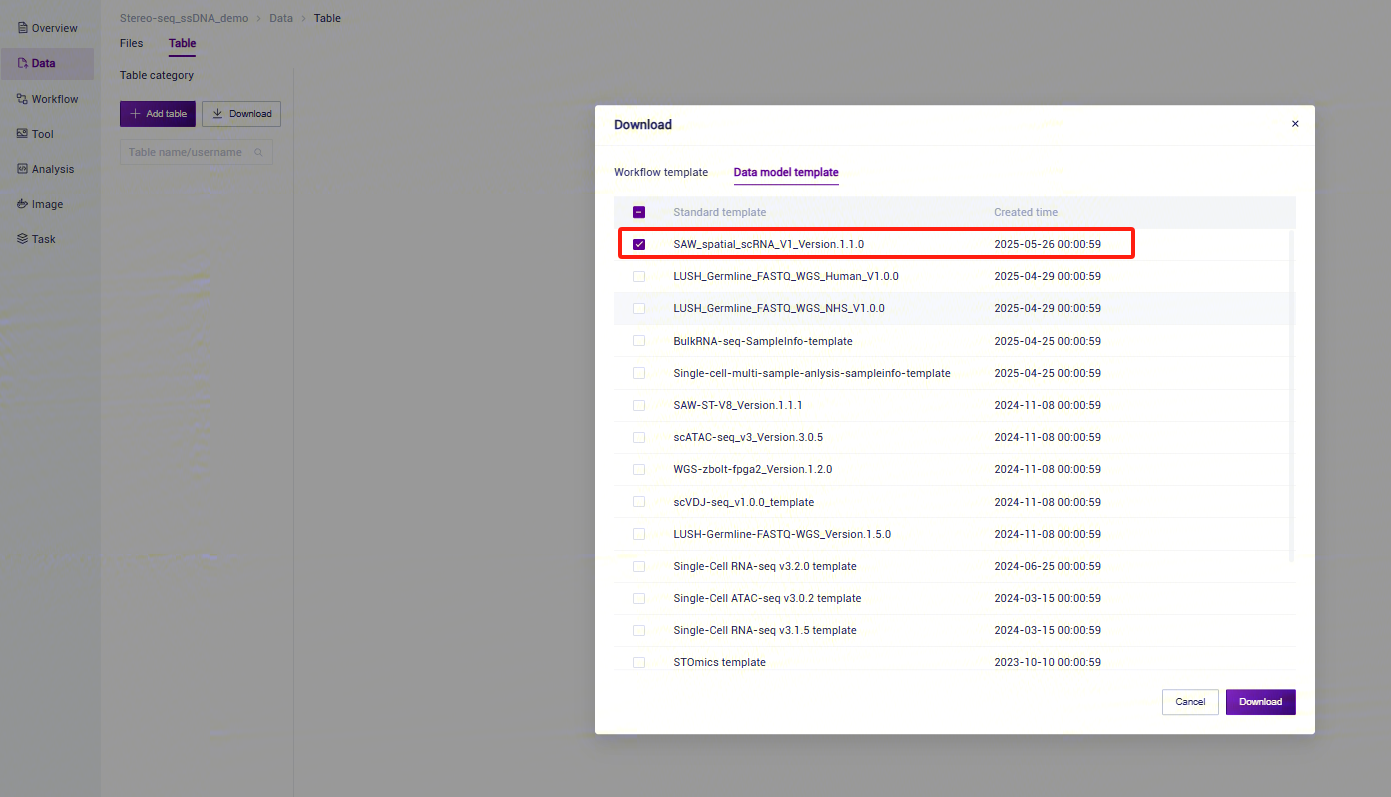
步骤三:样本信息录入表格下载
SAW_spatial_scRNA_V1
点击导航栏**【Data】,选择【Table】-【Download】(如图3-15), 点击【Workflow template】**,选择 模板下载:
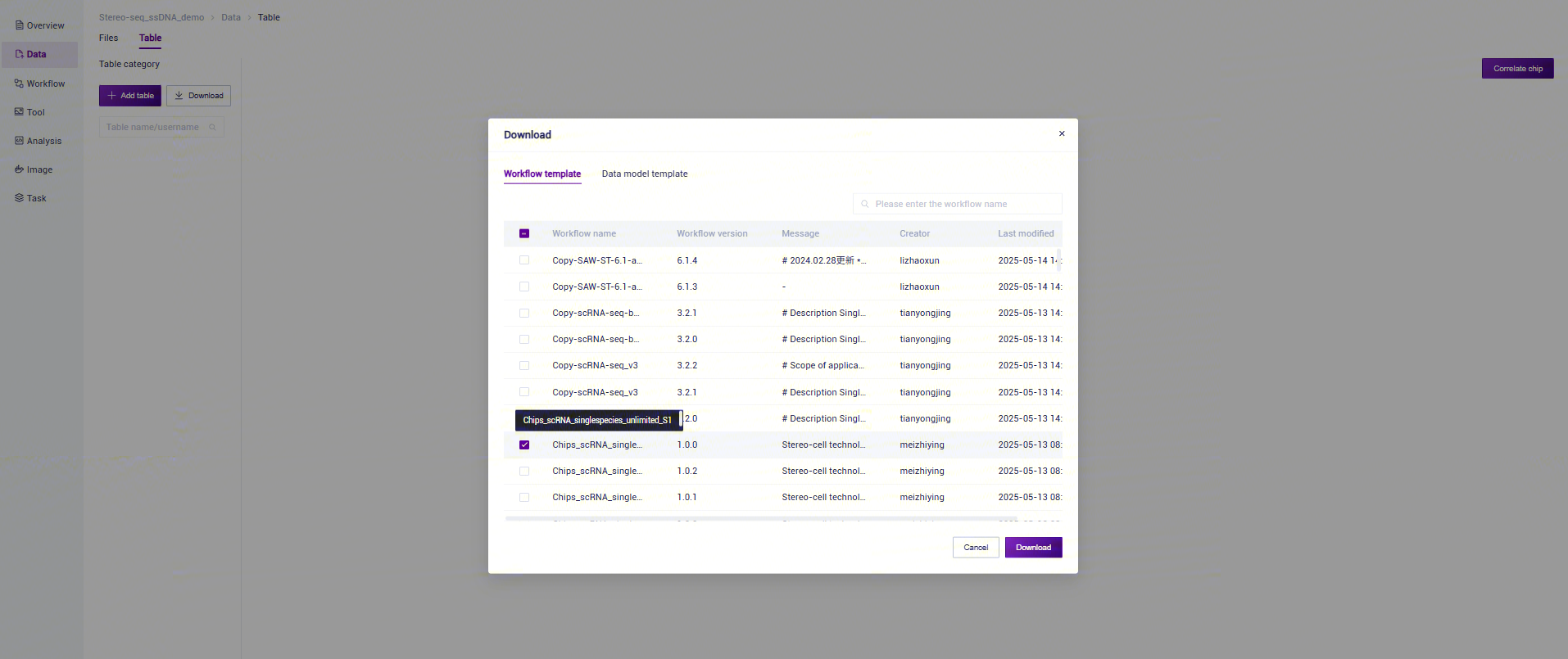
Chips_scRNA_singlespecies_unlimited_S1
点击导航栏**【Data】,选择【Table】-【Download】(如图3-16), 点击【Workflow template】**,选择 模板下载:
Chips_scRNA_singlespecies
点击导航栏**【Data】,选择【Table】-【Download】(如图3-17), 点击【Workflow template】**,选择 模板下载:
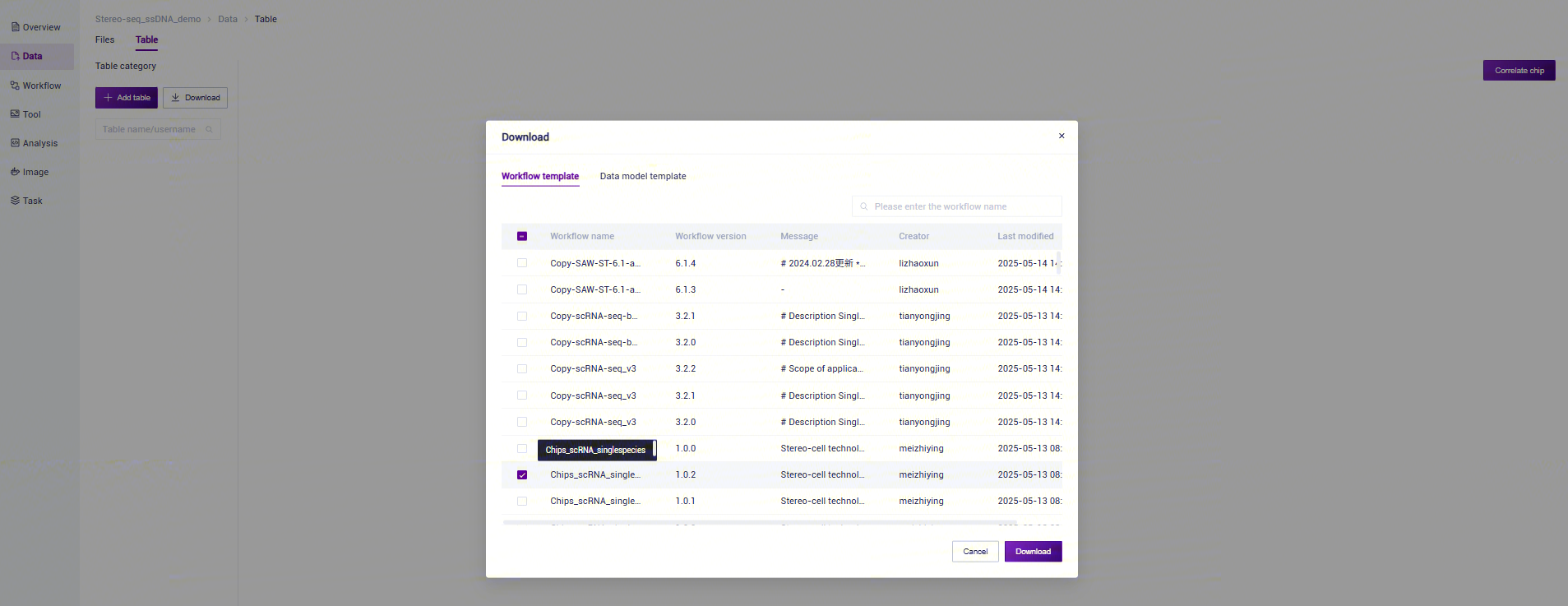
步骤四:样本信息导入
- 该使用场景条件下,样本导入表格需填写工作表,以**SAW_spatial_scRNA_V1****表格模板为例**(图3-15)。该场景表示对已测序完成的样本数据在导入表格后,直接进入分析。
注意事项:
[1] 导入文件路径必须在云平台已存在,若不存在文件,则不会生成任务。
[2] 模板中参数均为必填项五笔填写。导入数据中,必填项字段不得为空。
[3] Excel中的EntityID需唯一。
[4] Excel中不能合并单元格,单元格内容前后不能有空格或特殊字符。
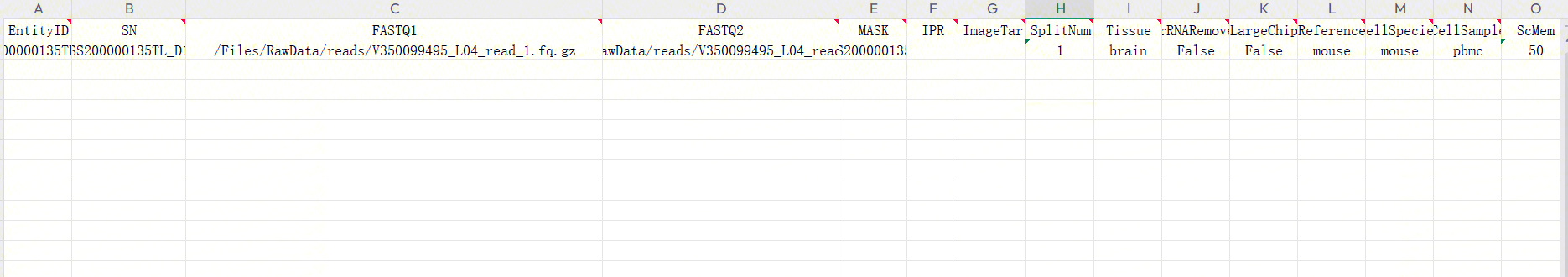
[5] 分析样本录入(图3-18):
同单样本信息内容录入方式:
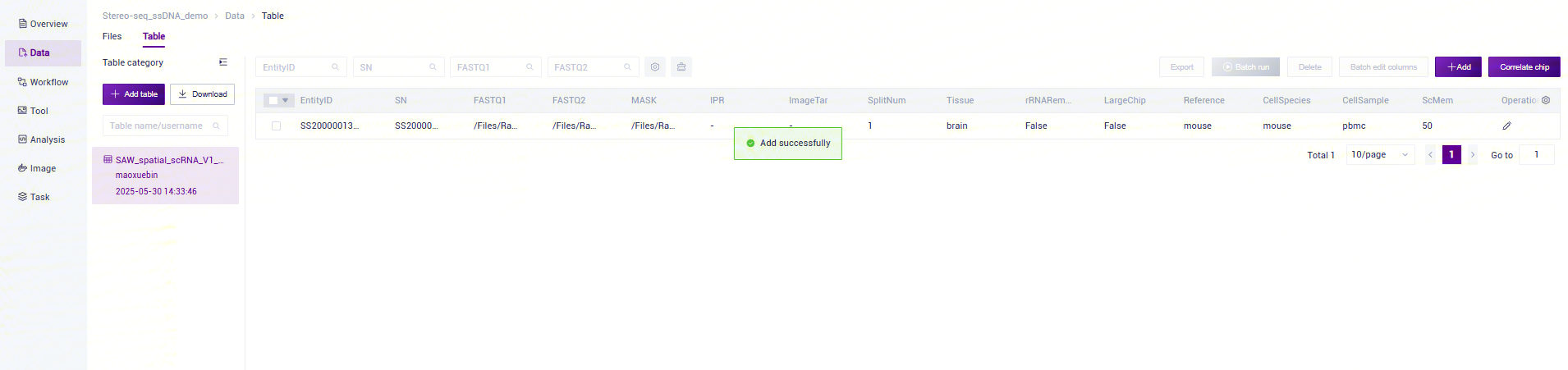
- 配置好样本模板的分析样本录入工作表后,回到**【Data】界面,点击【Table】-【+Add table】**(图3-19):
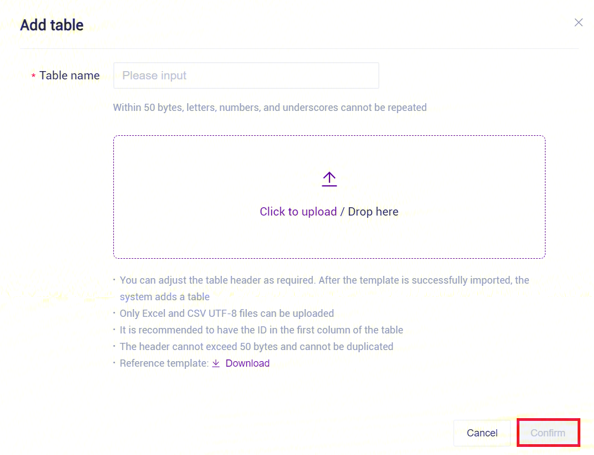
- 点击**【Click to upload/ Drop here】浏览并选择已填好样本信息的表格,点击【confirm】**(图3-20),上传完成后文件会在目标文件夹中显示:
- 点击导航栏**【Workflow】进入流程分析页面,在搜索框输入BulkRNA-seq****,点击【Run】**(图3-21):
- 选择Run workflow(s),点击 Please select table处,选择在本小节3)中导入的表格,选中所需的行,点击**【Next】**(图3-21):
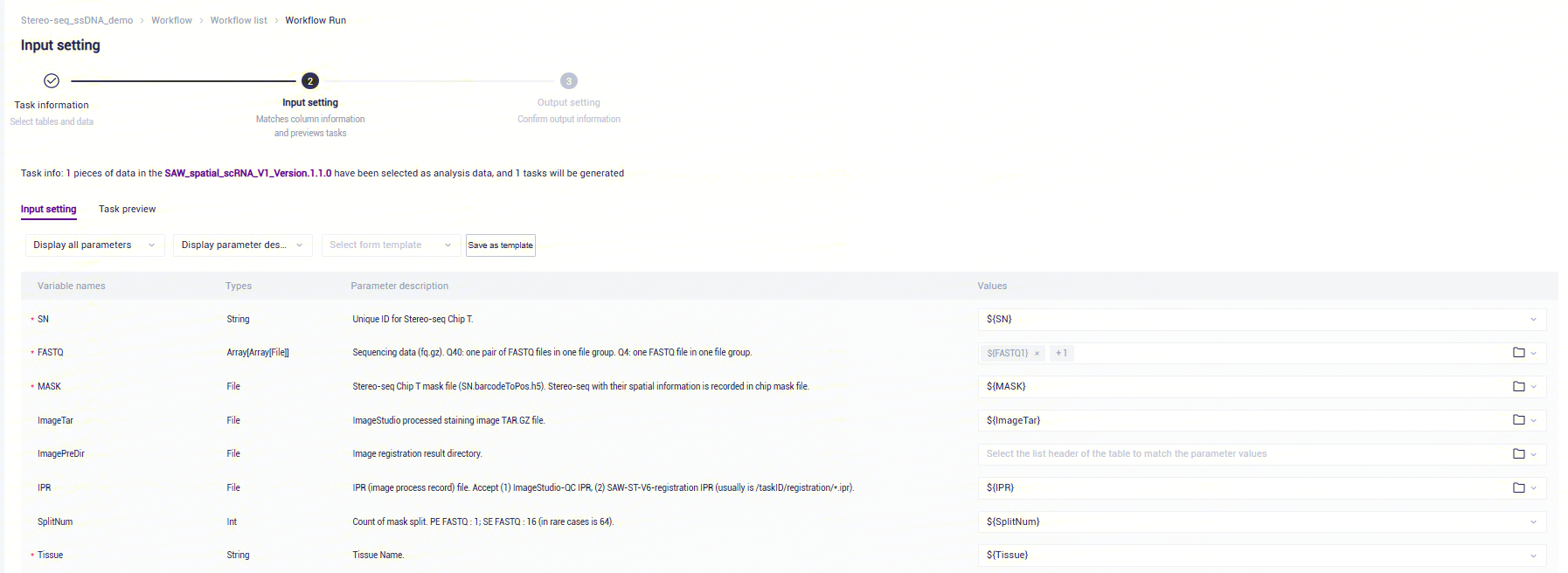
- Values一般会自动填充,仅需要检查填充是否正确,若需修改则在Values处点击并选择对应的值(如图3-23):
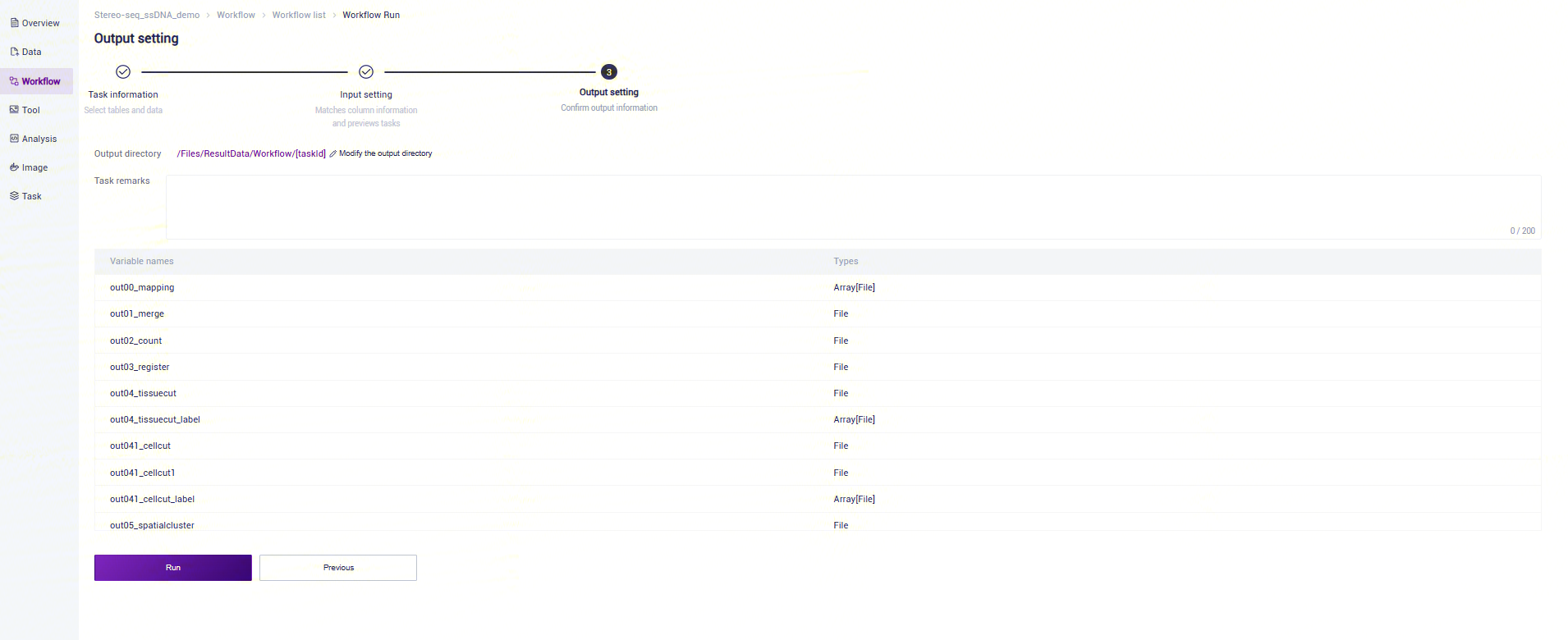
步骤五:启动分析
点击**【Run】**启动分析(图3-24):
报告查看
报告查看
点击导航栏**【Task】,当任务状态显示为completed时,代表任务已完成,点击【Detail】**即可查看结果部分(如图3-25):
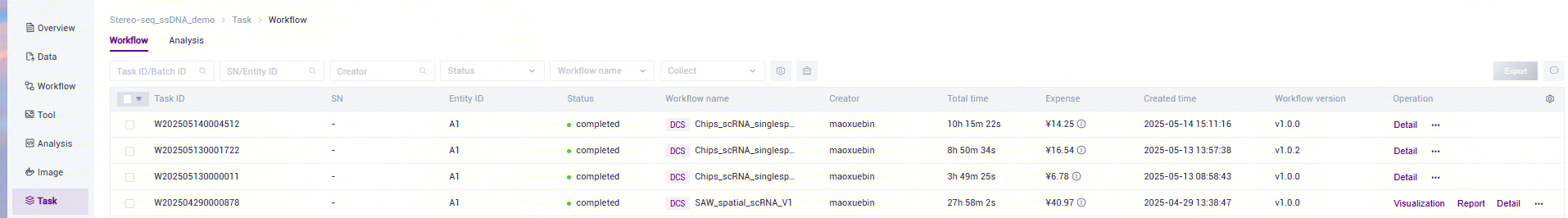
第四章 结果展示
报告&可视化展示
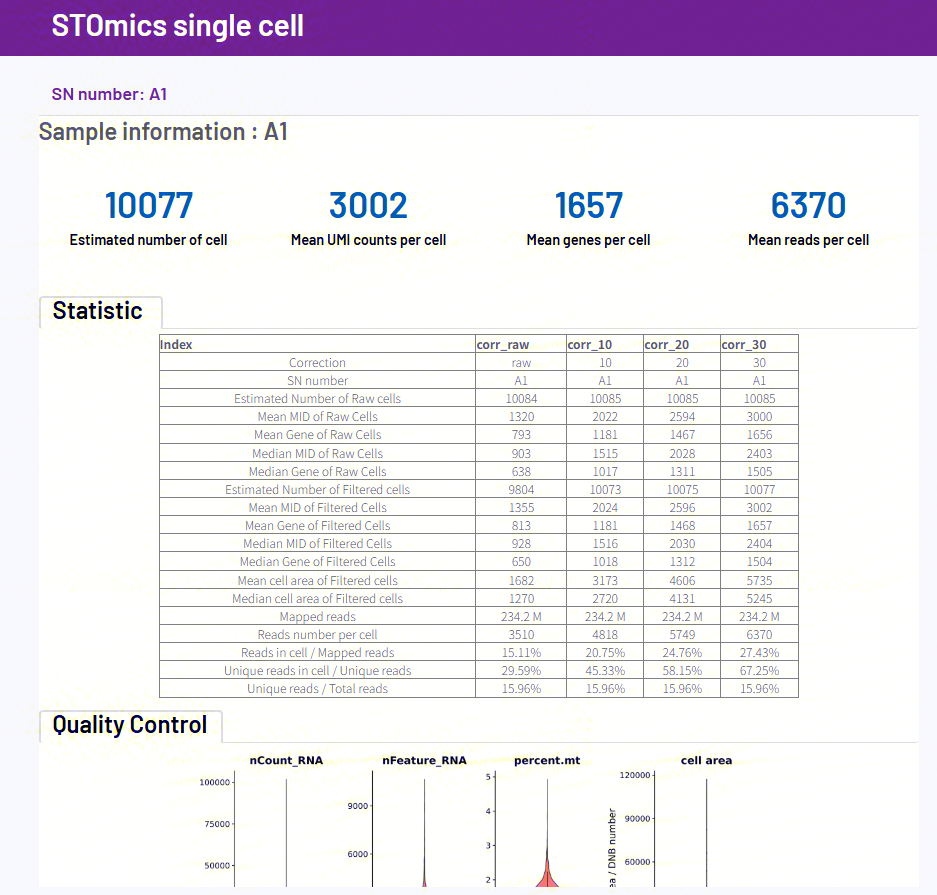
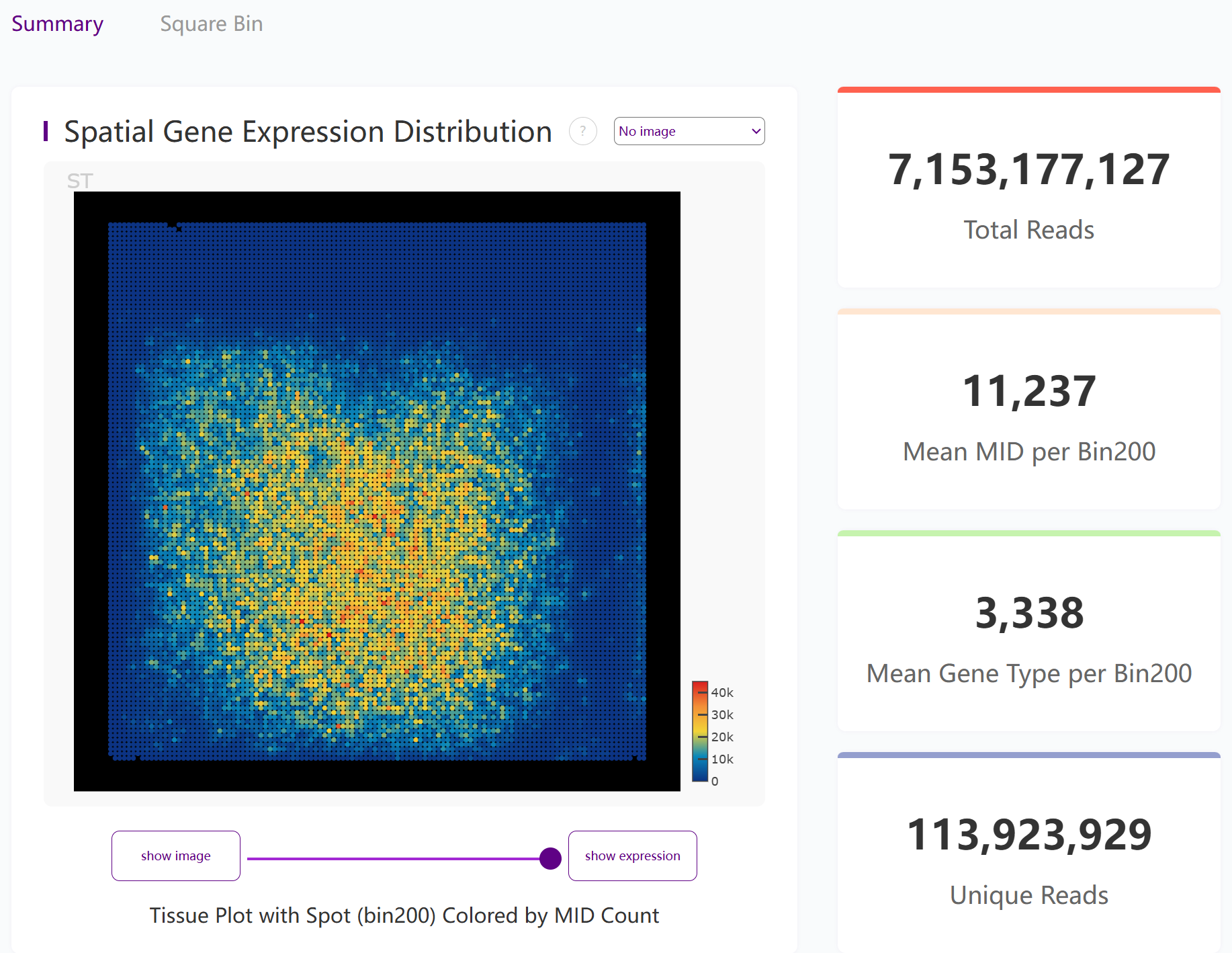
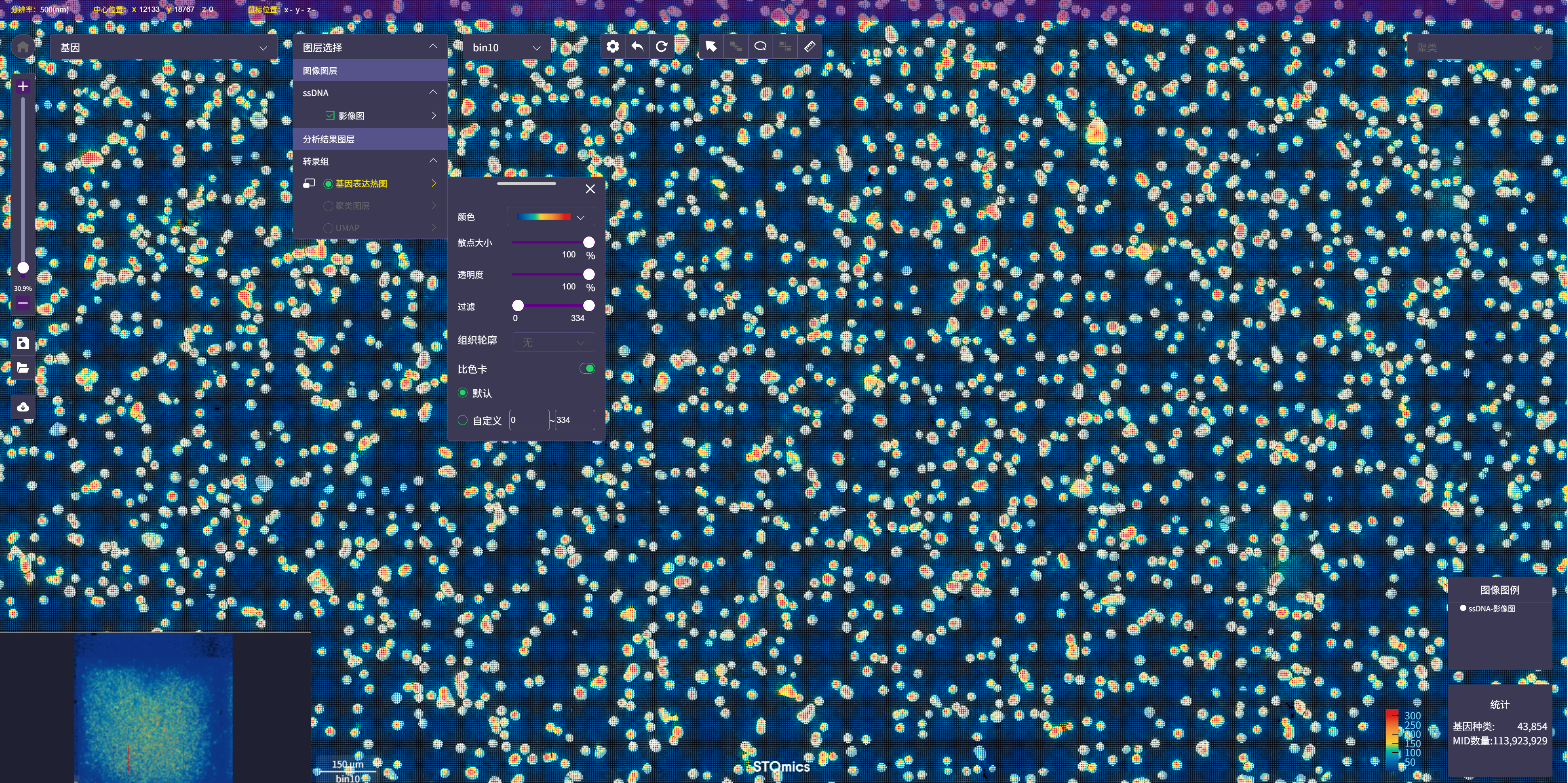
常见FAQ
Q: 申请要建ref,可以参考哪个资料说明?
A: 可以参考这个sop文档 https://alidocs.dingtalk.com/i/nodes/AR4GpnMqJzM3oe71CONRd01nVKe0xjE3?utm_scene=team_space