DCS Tools
1. Introduction
1.1 Description
DCS Tools is an advanced software leveraging CPU acceleration technology, designed to serve as an ideal alternative to BWA-MEM, GATK, and large-scale population joint calling in data analysis. It reduces storage costs by compressing Fastq.gz file sizes. Its functionalities include germline and Somatic mutation analysis for whole-genome sequencing (WGS) and whole-exome sequencing (WES), joint calling for population variant detection, and a compression/decompression tool for Fastq and vcf files used forgenomic data.
1.2 Advantages and Value
1)Consistent and reliable variant detection tool
This tool ensures the core algorithmic logic of modules like SOAPnuke, BWA, and GATK remains unchanged while incorporating excellent parallelization design and restructuring, accelerating computation and reducing unnecessary I/O operations. It not only significantly speeds up processing but also ensures high consistency with the GATK system.
Tool highlights:
Same math as the Broad Institute's BWA-GATK best practice workflow, but FASTQ to VCF is 16 times faster,Germline: 42 times faster from BAM to VCF, Somatic: BAM to VCF 8 times faster(Compared in units of nuclear-time)
No runtime variability, no downsampling in high-coverage regions.
Pure software solution running on CPU-based systems, cross-platform compatible with X86, ARM and cloud environments.
Tested on Alibaba Cloud ECS i4g.8xlarge.
- 30x genome, NA12878 (HG001), completed in 1.82 hours (58 core·hours).
2)Joint Calling: A tool for merging gVCFs and joint calling in population sequencing projects
Tool highlights
Fast: Completes joint variant detection for 52,000 whole genomes in 6 days. Accurate: GIAB standard sample SNP F1 score 99.44%, INDEL F1 score 98.95%. Scalable: Supports joint calling for millions of samples. User-friendly: Automatically allocates tasks, eliminating the need for users to design parallel strategies.
3)FASTQ format data compression tool
Follows the logic of most tools by independently compressing the ID, sequence, and quality values in FASTQ files. For sequence compression, in addition to entropy encoding, it provides sequence alignment modules based on HASH or BWT to improve the compression ratio for successfully aligned sequences. It also employs a data block design to balance compression ratio and performance, while the introduction of a packaging format ensures data security, independent partial data access, and backward compatibility.
Specialized Optimization
Maintains high compression and decompression speeds while ensuring a high compression ratio.
Reference genome sequences can exceed 4G.
Effective compression for both short and long reads.
Convenience and Flexibility
Supports Linux platforms, allows flexible setting of concurrent threads, and decompression has no license restrictions.
Complete Ecosystem
Seamlessly integrates with other tools in DCS Tools, enabling simultaneous decompression and computation.
Data is encapsulated in basic units, facilitating format extension and ensuring backward compatibility.
4)VCF format data compression tool
A tool for lossless compression of VCF(Variant Call Format) files generated by large-scale genome sequencing projects. The tool ensures high compression rates while achieving fast random access. Compress the vcf.gz file to 2/5 of the original file size, saving 60% of storage.
1.3 Platform Requirements
DCS Tools is designed to run as an executable program on Linux systems. Recommended Linux distributions include CentOS 7, Debian 8, Ubuntu 14.04, and later versions.
Hardware requirements for DCS Tools: •CPU: At least 8 cores. •Memory: At least 128G. •Recommended use of SSDs for faster read/write speeds.
1.4 License Verification
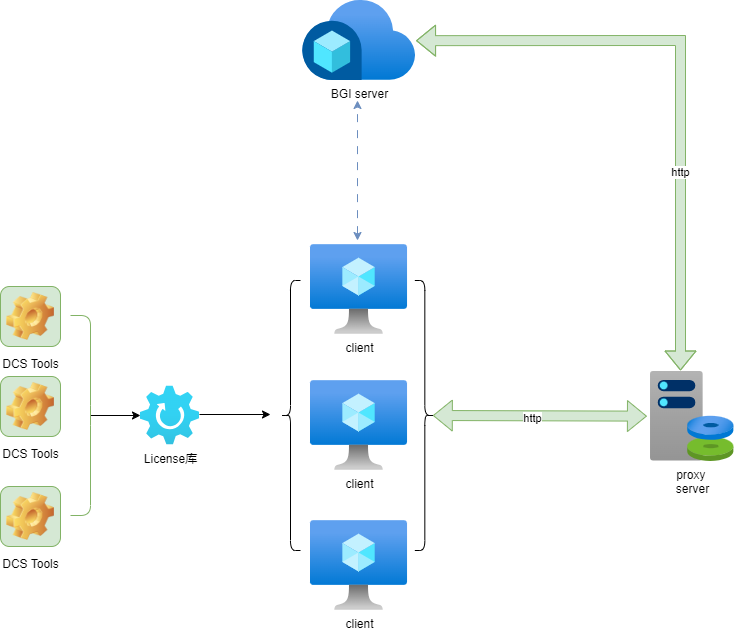
To verify the legality of the license during use, you need a compute node (also referred to as a "license node") to handle the verification communication. The compute node must ensure stable communication with the license server. Alternatively, this node can also serve as the verification reference (as shown in the example diagram at the end of this section). Additionally, you will need a license file, which can be obtained by contacting the platform (cloud@stomics.tech) for purchase or trial.
1.4.1 Obtaining the Compute Node's IP Address
Ensure that the compute node is not behind HTTP_PROXY or HTTPS_PROXY and can access cloud.stomics.tech.
When applying for a license file, you need to provide the external IP address of the compute node. You can obtain this IP address by running the following command:
curl --noproxy "\*" -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
The returned data field will contain the IP address:
{""code"":0,""msg"":""Success"",""data"":""172.19.29.215""}
If the following error occurs:
curl: (35) error:0A000152:SSL routines::unsafe legacy renegotiation disabled
Run the following command to obtain the IP address:
echo -e "openssl\_conf = openssl\_init\n\[openssl\_init\]\nssl\_conf = ssl\_sect\n\[ssl\_sect\]\nsystem\_default = system\_default\_sect\n\[system\_default\_sect\]\nOptions = UnsafeLegacyRenegotiation" | OPENSSL\_CONF=/dev/stdin curl --noproxy "\*" -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
1.4.2 Obtain the MAC Address of the Networked Node
When applying for a License file, you are required to provide the MAC address. Use the following command to retrieve the MAC address of the networked node:
./libexec/licproxy --print-mac
1.4.3 Obtaining the License File
Contact the platform at cloud@stomics.tech to purchase or request a trial license.
1.4.4 Deploying the License
After obtaining the license file, unzip the package and place the license file in the libexec directory.
1.4.5 Configuring the License Proxy on the Compute Node
On the compute node, run the foreground license proxy using the following command:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic
Replace /path/to/XXXXXXXXX.lic with the actual path to your license file. The license proxy defaults to listening on 0.0.0.0:8909. If this address is already in use, you will see a warning:
"\[2025-03-27T06:34:46Z WARN pingora\_core::listeners::l4\] 0.0.0.0:8909 is in use, will try again"
The system will automatically retry, or you can manually stop the process with Ctrl + C and specify a different address:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic --bind 0.0.0.0:8910
If no errors occur, the proxy will start successfully:
\[2025-03-27T07:15:52Z INFO pingora\_core::server\] Bootstrap starting
\[2025-03-27T07:15:52Z INFO pingora\_core::server\] Bootstrap done
\[2025-03-27T07:15:52Z INFO pingora\_core::server\] Server starting
Before proceeding, verify that the license proxy can communicate with the platform:
curl http://127.0.0.1:8909/api/licensor/licenses/anon/pool
The expected response indicates successful communication:
{""code"":0,""msg"":""Success""}
To run the proxy in the background, use the -d flag:
./libexec/licproxy --license /path/to/XXXXXXXXX.lic -d
This will run the proxy as a daemon process.
Note: Only one license proxy is needed per node when running in the background. Multiple license proxies are supported for the same Linux user on the same node, but not for multiple Linux users on the same node. To stop the proxy, use kill or delete the file /tmp/pingora.pid.
1.4.6 Running DCS Tools
After completing the above steps, the compute node can communicate with the license node to run DCS Tools. For processing WGS sequencing data, you can use the quick_start script provided by the platform. Refer to the @example demonstration script for details.
Before running, set the PROXY_HOST environment variable to the IP and port of the license proxy. Ensure that the current node can ping the IP. If the compute node and license node are the same machine, use 127.0.0.1 as the IP.
To verify the connection between the compute node and license node:
$DCS\_HOME/bin/dcs lickit --query $PROXY\_HOST
To compress a FASTQ file using DCS Tools:
export PROXY\_HOST=<ip>:<port>
$DCS\_HOME/bin/dcs SeqArc compress -i input.fastq.gz -o output.fastq
1.4.7 Steps After Upgrading
After obtaining an upgraded version, follow the instructions provided. Generally, you do not need to reconfigure the license proxy (version 1.4.4). Simply unzip the new package and update the DCS_HOME path to the new directory.
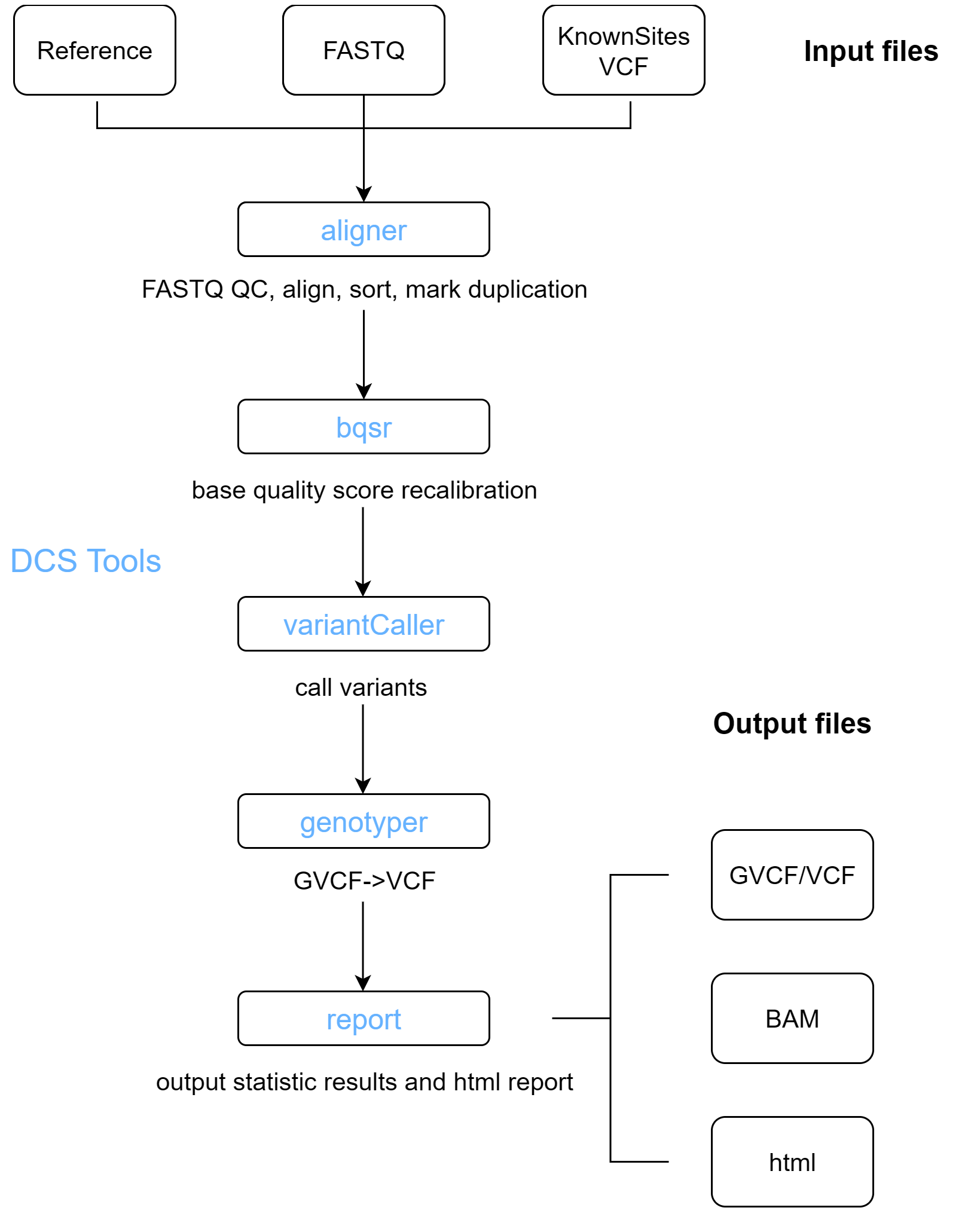
1.5 Tool List
| Scenario | Tool (Module) Name | Function |
|---|---|---|
| WGS/WES Analysis | aligner | Quality control, alignment, sorting, and duplicate marking |
| bqsr | Base quality score recalibration | |
| variantCaller | Variant detection (similar to GATK HaplotypeCaller) | |
| genotyper | GVCF->VCF | |
| report | Output report | |
| Joint Calling for Large Cohorts | jointcaller | Joint variant detection, supports millions of samples |
| FASTQ Data Compression/Decompression | SeqArc | High-efficiency lossless FASTQ compression tool |
| VCF data compression decompression | VarArc | Efficient lossless VCF compression tool |
| License Query, etc. | lickit | Query available threads, test network connection, upload error logs, etc. |
| Utility Tools | extract-vcf | Simplify VCF files, reduce file size, alleviate I/O pressure, primarily for knownSites (e.g., dbSNP) input for bqsr and genotyper |
| bam-stat | Alignment statistics | |
| vcf-stat | Variant statistics | |
| tbi2lix | Convert TBI index files to custom linear index files for joint calling |
2. Typical Usage
2.1 WGS
WGS (Whole Genome Sequencing) is a technology that sequences the entire genome of an organism. The WGS analysis pipeline typically includes steps such as data quality control, sequence alignment, duplicate marking, base quality score recalibration, and variant detection. DCS Tools follows the original algorithms while optimizing performance, resulting in a 16x faster analysis pipeline compared to BWA-GATK.
2.1.1 Preparations
Input files for the WGS analysis pipeline:
- Files required for sequence alignment:
| Index File | Description |
|---|---|
| species.fa | Reference genome FASTA file |
| species.fa.fai | For rapid location and random access to FASTA file regions |
| species.dict | Index for quick lookup of reference sequences in subsequent processes |
| species.fa.* | Alignment index files |
Build alignment indexes using the aligner tool. Place the index files in the same directory as the FASTA file. The command to build alignment indexes is:
dcs aligner --threads <INT> --build-index <FASTA>
- FASTQ sequencing data, supports gz format.
- Single nucleotide polymorphism (dbSNP) data and known variant sites in VCF format.
The WGS analysis pipeline provided by DCS Tools includes the following steps:
- Sequence alignment and duplicate marking: Quality control is performed before alignment. The sequences are aligned to the reference genome recorded in the FASTA file, and the alignment results are written to a BAM file after duplicate marking.
- Base quality score recalibration: Adjusts the quality scores assigned to each base in the sequence to eliminate experimental biases introduced by sequencing methods.
- Variant detection: Identifies variant sites in the sequencing data relative to the reference genome and calculates genotypes for each sample at these sites.
2.1.2 Sequence Alignment and Duplicate Marking
Align sequences to the reference genome to determine the position of each sequence, providing a foundation for variant detection. Duplicate marking eliminates PCR-amplified duplicate sequences, reducing false positives and improving variant detection accuracy. These steps are completed by the aligner tool. The aligner also integrates data quality control functionality, filtering and processing sequences before alignment to reduce error rates and avoid interference from noisy data in subsequent analyses.
dcs aligner --threads THREADNUM \
--aln-index INDEX \
--fq1 FASTQ1 \
--fq2 FASTQ2 \
--read-group RGROUP \
--bam-out BAMFILE \
--fastq-qc-dir QCDIR \
--high-speed-storage TEMPDIR
The command requires the following inputs:
- THREADNUM: Number of threads. It is recommended not to exceed the number of CPU cores.
- INDEX: Path to the reference genome FASTA file. Place the alignment index files built by the alignment software in the same directory as the FASTA file.
- FASTQ1: Path to the FASTQ1 file for paired-end sequencing.
- FASTQ2: Path to the FASTQ2 file for paired-end sequencing.
- RGROUP: Format is @RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM, where GROUP_NAME is the read group identifier, SAMPLE_NAME is the sample name, and PLATFORM is the sequencing platform.
- BAMFILE: BAM file after duplicate marking.
- QCDIR: Output location for the quality control report.
- TEMPDIR: Storage path for temporary files. Placing this path on high-speed storage devices can improve software performance.
2.1.3 Base Quality Score Recalibration
Due to systematic errors, the base quality scores of sequences are not always entirely accurate. Recalibration corrects these biases, generating more precise quality scores to improve variant detection accuracy and reduce false positives and negatives. This step aligns with GATK's best practices and is completed by the bqsr tool.
dcs bqsr --threads THREADNUM \
--in-bam BAMFILE \
--reference FASTA \
--known-sites KNOWN_SITES \
--recal-table RECAL_TABLE
The command requires the following inputs:
- THREADNUM: Number of threads. It is recommended not to exceed the number of CPU cores.
- BAMFILE: Input file. The BAM file generated after sequence alignment and duplicate marking.
- FASTA: Path to the reference genome FASTA file. Ensure it is the same as the one used in the sequence alignment stage.
- KNOWN_SITES: Single nucleotide polymorphism database data and known variant sites file.
- RECAL_TABLE: Output file. Saves information for recalibrating base quality scores.
2.1.4 Variant Detection
Detects SNP and INDEL variants, generates a GVCF file, and produces BAM statistics. The statistics file is stored in the same directory as the GVCF file, named bamStat.txt. This step is completed by the variantCaller tool.
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-t THREADUM
The command requires the following inputs:
- INPUT_BAM: The BAM file output from step 2.1.2 (sorted and duplicate-marked).
- OUTPUT_GVCF: Output GVCF file.
- REFERENCE: Reference genome FASTA file.
- RECAL_TABLE: Input file containing information for base quality score recalibration, generated in step 2.1.3.
- THREADNUM: Number of threads. It is recommended not to exceed the number of CPU cores.
For a single sample, if only a VCF file containing variant records is needed, the genotyper tool can be used.
dcs genotyper -I INPUT_GVCF \
-O OUTPUT_VCF \
-s QUAL \
-t THREDNUM
The command requires the following inputs:
- INPUT_GVCF: GVCF file from variantCaller.
- OUTPUT_VCF: Output VCF file.
- QUAL: Variant quality score threshold. Records with quality scores greater than or equal to QUAL will be output.
- THREADNUM: Number of threads. It is recommended not to exceed the number of CPU cores.
2.1.5 Report Generation
Consolidates statistics from each sample to generate a visual HTML report.
dcs report --sample SAMPLE_NAME \
--filter QCDIR \
--bam_stat BAMStat \
--vcf_stat VCFStat \
--output OUTPUT_DIR
The command requires the following inputs:
- SAMPLE_NAME:Sample name.
- QCDIR: Quality control statistics report folder generated by the aligner.
- BAMStat: bamStat.txt in the variantCaller result directory.
- VCFStat: VCF statistics results, defaulting to vcfStat.txt in the working directory.
- OUTPUT_DIR: Report output directory.
2.2 WES
The steps are largely the same as WGS, with the only difference being in the variantCaller step, which requires an additional interval parameter (-L).
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-L INTERVAL_FILE
-t THREADUM
INTERVAL_FILE: BED file recording WES intervals. By convention, each line follows the format: chr<tab>start<tab>end Where start and end positions are 0-based and left-closed, right-open intervals.
2.3 Joint Calling
Joint Calling is a method for detecting population variants, specifically referring to the process of merging variants from multiple samples and recalculating sample genotypes using multiple GVCF files as input. DPGT is a distributed Spark program that efficiently performs joint calling for large-scale samples. dcs jointcaller is a wrapper for the DPGT tool, used to run DPGT in standalone mode.
# Set JAVA_HOME to jdk8
export JAVA_HOME=/path_to/jdk8
# Add spark bin to PATH for calling spark-submit
export PATH=/path_to/spark/bin/:$PATH
dcs jointcaller \
-i GVCF_LIST \
-r REFERENCE \
-o OUTDIR \
-j JOBS \
--memory MEMORY \
-l REGION
DPGT depends on Java 8. Set JAVA_HOME=/path_to/jdk8. Spark version 2.4.5 or higher is recommended. Download Spark 2.4.5 from: https://archive.apache.org/dist/spark/spark-2.4.5/spark-2.4.5-bin-hadoop2.6.tgz
The command requires the following inputs:
GVCF_LIST: Input GVCF list, a text file with one GVCF path per line. Note that GVCFs must have matching TBI index files. The program defaults to searching for TBI index files by appending .tbi to the GVCF path.
REFERENCE: Path to the reference genome FASTA file. Ensure this FASTA file is consistent with the one used to generate the GVCFs. The program also requires matching .dict and .fai files for the FASTA file (e.g., if the FASTA file is hg38.fasta, the program will also read hg38.dict and hg38.fasta.fai).
OUTDIR: Output folder. The resulting VCF path is OUTDIR/result.*.vcf.gz.
JOBS: Number of parallel tasks. The number of threads used is equal to JOBS, an integer ranging from 1 to the number of machine threads.
MEMORY: Maximum Java heap memory size. The unit is "g" or "m". Memory size is related to the JOBS value and sample size. For 1,000 samples, allocate 2g per job; for 2,000-5,000 samples, allocate 4g per job; for 5,000-100,000 samples, allocate 6g per job; for 100,000-500,000 samples, allocate 8g per job.
REGION: Target region. Supports input strings or BED paths. The region string format is consistent with bcftools, e.g., "chr20:1000-2000", "chr20:1000-", "chr20". To specify multiple regions, use multiple -l parameters, e.g., -l chr1 -l chr2:1000-2000.
2.4 FASTQ compression
The SeqArc module is used to efficiently compress and decompress FASTQ data. This module optimizes the compression strategy for the second generation of short-read and the third generation of long-read sequencing data, and supports three different compression modes:
- No reference mode: does not rely on any Reference File, compression speed is faster, compression ratio is lower.
- Single reference mode: Uses a specified reference genome (FASTA) to improve compression ratio and consistency during compression/decompression.
- Multi-reference mode: provides multiple reference genomes, and the program automatically matches the most suitable single FASTA for compression and decompression.
Compared with general-purpose compression tools, the SeqArc module has significant advantages in storage efficiency and decompression speed, especially for large-scale sequencing data archiving and transmission scenarios.
2.4.1 Environment and Input/Output Requirements
2.4.1.1 Requirements for files to be compressed
FASTQ file, which supports uncompressed text format (.fq or. fastq) and gz compressed format (.fq.gz or .fastq.gz).
2.4.1.2 Reference Document Requirements
If single-reference mode or multi-reference mode is used, a reference genome FASTA file is required.
If multiple reference mode is used, there should be multiple FASTA files in the Index Directory.
Needs to download and decompress the NCBI taxonomy packagetaxdump.tar.gz(http://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz) To this directory (accounting for 485MB of space) to support reference genome matching.
2.4.1.3 Compressed Output File Format
After compression:.arcFile.
After decompression: FASTQ or FASTQ.gz File (gz can specify the compression level).
2.4.2 Index construction
2.4.2.1 Usage (Usage)
dcs SeqArc index [options] -r /path/to/ref.fa
dcs SeqArc index [options] -K /path/to/ref_dir/
2.4.2.2 Function description
Reference genomes are indexed to support "single-reference compression" and "multi-reference compression". If you specify-K(Multiple reference FASTA directory), will build a classification database, so that the compression stage can automatically select the most appropriate reference, decompression stage can also automatically select the reference used in compression. Using the "dcstools SeqArc index -r REF -o OUTDIR" command will generate two indexes for compression, namely, the second-generation short-read index and the third-generation long-read index. Taking the human genome as an example, GRCh38.fa is 3.1GB,It takes about half an hour to build the index. The output of the second generation short read long index GRCh38.fa. Mini_28_10, accounting for 14GB, the output of the third generation long read long index GRCh38.fa. Similar_26_23, accounting for 6.1GB,+ + The peak memory in the process is about 35GB,For FASTA larger than the human genome, memory consumption increases proportionally.
2.4.2.3 Parameter Description
| Parameters | Explain | Default Values/Key Points |
|---|---|---|
-r, --ref <file> | Specify a reference FASTA file | Necessary for single reference mode |
-K, --clsdb <dir> | Specifies a directory containing multiple reference FASTA | For species identification patterns |
-H, --hitlimit <int> | Maximum number of matches when reference matches | Default 2 |
-t, --thread <int> | Number of threads used | Default 8 |
-o, --output <dir> | Index Output Directory | Use current directory or default subdirectory if not specified |
-h, --help | Help show this message | - |
2.4.3 Compression
2.4.3.1 Usage (Usage)
dcs SeqArc compress [options] -i reads.fq -o reads.arc
dcs SeqArc compress [options] -r /path/to/ref.fa -i reads.fq -o reads.arc
dcs SeqArc compress [options] -K /path/to/ref_dir/ -i reads.fq -o reads.arc
2.4.3.2 Function description
Compress the FASTQ file.arcFormat.
Three compression modes are available: no reference, single reference, and multiple reference.
It is worth noting that although multiple fastas are used in the multi-reference mode, only the one closest to the current FASTQ is tried from the fastas of multiple databases (or the no-reference mode is selected because none of them match), and only one FASTA file is used as a reference in the compression and decompression process.
2.4.3.3 Parameter Description
| Parameters | Explain | Default/Recommended |
|---|---|---|
-i, --input <file> | Input FASTQ file | Required |
-o, --output <file> | Output file, will increase the decision, to ensure that the output file suffix is.fq.arcOr.fastq.arc | Required |
-r, --ref <file> | Single reference FASTA | Optional |
-K, --clsdb <dir> | Multi-reference catalogs for species identification | Optional |
-H, --hitlimit <int> | Reference match upper limit | Default 2 |
-J, --clsmin <float> | Minimum Read hit ratio | Default 0.6 |
-L, --clsnum <int> | Number of reading samples for classification judgment | Default 10000 |
-t, --thread <int> | Number of threads | Default 8 |
-f, --force | Overwrite existing output file | - |
--pipein | Read input from pipe | Suitable for streaming |
--inputname <str> | Specifies the original FASTQ name only if you use--pipeinThis name is used when decompressing without specifying an output file name. | - |
--blocksz <size> | Block size in MB | Default 50 |
--ccheck | Decompress the check content immediately after compression, slow but safe | - |
--calcmd5 | Calculate the MD5 checksum of the input FASTQ | For subsequent verification |
--slevel <int> | Sequence compression level (8-15) | Default 10 |
--verify <int> | Verification mode: 0 (none),1 (whole block),2 (whole block + name, sequence, quality verification respectively) | Default 1 |
-h, --help | Help show this message | - |
2.4.4 Decompression
2.4.4.1 Usage (Usage)
dcs SeqArc decompress [options] -i reads.arc -o reads.fq
dcs SeqArc decompress [options] -r /path/to/ref.fa -i reads.arc -o reads.fq
dcs SeqArc decompress [options] -K /path/to/ref_dir/ -i reads.arc -o reads.fq
2.4.4.2 Function description
Will.arcUnzip the file to a FASTQ file.
If a reference or species identification library is used during compression, the corresponding reference or reference library should also be provided during decompression to ensure correct recovery of the sequence.
2.4.4.3 Parameter Description
| Parameters | Explain | Default/Recommended |
|---|---|---|
-i, --input <file> | Input.arcFile | Required |
-o, --output <file> | Output FASTQ file path (output to standard output if not specified) | - |
-r, --ref <path> | Single reference FASTA file or directory | If you use-rPattern |
-K, --clsdb <dir> | Multi-reference catalogs for species identification | If the compression is used-KPattern |
-t, --thread <int> | Number of threads | Default 8 |
-f, --force | Overwrite existing output file | - |
--type <int> | Output type: 0 = gz compressed FASTQ;1 = plain fastq text | Default 0 |
--gzlevel <int> | gzip compression level | Default 4 |
--dcheck | Read and verify again after writing | Slower but more reliable |
--show | If compression is used--calcmd5Displays the MD5 value of the original FASTQ | - |
-h, --help | Help show this message | - |
2.4.5 Inspection
2.4.5.1 Usage
dcs SeqArc check [options] -i reads.arc
dcs SeqArc check [options] -r /path/to/ref.fa -i reads.arc
dcs SeqArc check [options] -K /path/to/ref_dir/ -i reads.Bow
2.4.5.2 Function description
Check.arcWhether the file is completely intact.
If a reference or species identification library is used for compression, the corresponding reference or reference library should also be provided for inspection.
2.4.5.3 Parameter Description
| Parameters | Explain | Default/Recommended |
|---|---|---|
-i, --input <file> | Input.arcFile | Required |
-r, --ref <path> | Single reference FASTA file or directory | If the compression is used-rPattern |
-K, --clsdb <dir> | Multi-reference catalogs for species identification | If the compression is used-KPattern |
-t, --thread <int> | Number of threads | Default 8 |
-f, --force | Overwrite existing output file | - |
-h, --help | Help show this message | - |
2.4.6 Output statistics
2.4.6.1 Usage (Usage)
dcs SeqArc stat [options] -i reads.arc
2.4.6.2 Function description
Output.arcFile statistics in the compression process to get the data.
2.4.6.3 Parameter Description
| Parameters | Explain | Default/Recommended |
|---|---|---|
-i, --input <file> | Input.arcFile | Required |
-h, --help | Help show this message | - |
2.4.7 Parameter selection and use recommendations
| Status | Suggest mode | Key parameter tuning |
|---|---|---|
| The amount of data is very large, and the maximum compression rate is expected. | Single Reference (-r) or multi-reference (-K) Mode | Goose--slevel, use more threads; Confirm that the reference genome matches the sample species well |
| Mixed data types, unknown species | Multi-reference mode | Prepare several reference FASTA and download taxonomy data; Adjust-L,-clsminEnsure accurate classification |
| Requires fast speed, the compression rate is not high | No reference mode | Use the default compression level, no.-rOr-K; Reduce verification options (e. G. Off--ccheck,--verify) |
| Hope that the compressed and decompressed data is exactly the same | Turn on the verification option | Use--ccheck(Check after compression), use--dcheck(Decompression verification), save the MD5 verification value |
2.4.8 Example
2.4.8.1 Index construction
First, you need to set the handle of the file system.
# Choose one of the two # Only take effect temporarily for the current session. If you need to take effect for a long time, you need to add it to the environment variable. ulimit -n 65535# set soft limit to 65535 ulimit -Hn 65535# Set hard limit to 65535 (requires root permission)For the single-reference case, create the index file of the reference sequence directly
dcs SeqArc index -r REF -o OUTDIRFor the multi-reference case, create a species identification library index and an index file for each species
# I. Create a new multi-species File Directory
mkdir fasidx && cd fasidx
# ii. Download the species relationship file and unzip it
wget http://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
tar xzf taxdump.tar.gz
# iii. Download the required FASTA species into the/path/to/fa directory and decompress fa.gz into fa
gunzip ./*fa.gz
# iv.Create the species identification library index clsf.cdb,clsf_del.cdb,clsf_opt.cdb,clsf_tax.cdb
dcs SeqArc index -K.
# v. Create a corresponding alignment index for each species (e. G. aaa.fa)
dcs SeqArc index -r aaa.fa
2.4.8.2 Use dcs for compression
- If the user knows the species of fastq, he can use the [-r] parameter to directly select the reference sequence of the corresponding species for compression:
dcs SeqArc puresss -r REF \ -I FASTQ \ -O ARC \ -T THREADIf the user does not know the specific species, you can use the species identification library for parameter compression, the program will use the species identification library to analyze the species of the input data, and then select the corresponding index file for parameter compression.
dcs SeqArc compress -K REFDIR \ -I FASTQ\ -O ARC\ -T THREADNUMEIf the user does not have a reference sequence, it can also be compressed without parameters.
dcs SeqArc puresss -i FASTQ \
-O ARC \
-T THREAD
The command requires the following parameters to be entered:
- REF: Input reference sequence path
- REFDIR: a directory containing multi-species reference sequences
- FASTQ: Enter the path to the fastq file
- ARC: output compressed file path
- THREADNUM: Number of threads set
2.4.8.3 Decompress using dcs
If the user knows the specific reference sequence used for compression, he can use the [-r] parameter to directly select the reference sequence of the corresponding species for compression.
Dcs SeqArc decompress -r REF \ -I ARC \ -O FASTQ \ -T THREADIf you use the species identification library for compression, you need to specify the path of the species identification library during decompression.
dcs SeqArc decompress -r REFDIR\ -I ARC\ -O FASTQ\ -T THREADNUMEIf the user is no-parameter compression, decompression is also no-parameter decompression.
dcs SeqArc decompress -i ARC \
-O FASTQ \
-T THREAD
The command requires the following parameters to be entered:
- REF: Input reference sequence path
- REFDIR: a directory containing multi-species reference sequences
- FASTQ: output fastq file path
- ARC: Enter the compressed file path
- THREADNUM: Number of threads set
2.4.8.4 Decompression using a separate decompression tool
If the customer does not purchase DCS Tools and only needs to decompress files in arc format, he can decompress the data with an independent free decompression tool, and obtain the decompression tool software package (Seqarc_decompress) from sales.
If the user knows the specific reference sequence used for compression, he can use the [-r] parameter to directly select the reference sequence of the corresponding species for compression.
SeqArc_decompress -r REF \ -I ARC\ -O FASTQ\ -T THREADNUMEIf you use the species identification library for compression, you need to specify the path of the species identification library during decompression.
SeqArc_decompress -r REFDIR\ -I ARC\ -O FASTQ\ -T THREADNUMEIf the user is no-parameter compression, decompression is also no-parameter decompression.
SeqArc_decompress -i ARC \
-O FASTQ\
-T THREADNUME
The command requires the following parameters to be entered:
- REF: Input reference sequence path
- REFDIR: a directory containing multi-species reference sequences
- FASTQ: output fastq file path
- ARC: Enter the compressed file path
- THREADNUM: Number of threads set
2.4.9 Precautions
- If used in compression-rOr-KMode, the corresponding reference or directory must be provided during decompression to ensure the integrity and consistency of the content.
- Multithreading can speed up, but resource (CPU + memory) constraints can cause performance bottlenecks.
- Higher compression levels (--slevelLarge) and verification options can significantly increase runtime.
- File Overwrite (-f, --force) Use with caution to avoid overwriting important data by mistake.
- In use--pipeinOr read from standard input, it is recommended to specify--inputnameIt is used to correctly identify the input file name and avoid unclear file name after decompression.
VarArc
VarArc compression compresses the common VCF text or VCF.gz file into smaller files to save storage space, reduce data transmission time, and improve processing efficiency.
The compression command is as follows:
Dcs Varc
-O ARC \
-T THROUGH \
--Rowcnt RCNT \
--Blkcolcnt BCNT
The command requires the following parameters to be entered:
VCF: Entered vcf file path
ARC: output compressed file path
THREADNUM: Number of threads set
RCNT: sets the number of rows, each RCNT is a large block.
BCNT: set the number of sample columns, each BCNT is a small block
The decompression command is as follows:
dcs VarArc decompress -i ARC \
-o VCF \
-t THREADNUM \
-p POSRANGE \
-s SAMPLELIST
The command requires the following parameters to be entered:
ARC: Enter the compressed file path
VCF: Path of the output unzip vcf file
THREADNUM: Number of threads set
POSRANGE: Set the chromosome name and interval of the query [chr1: 1000-2000]]
SAMPLELIST: Set the sample command list file for query [each line of the file is a sample]]
view can decompress and view vcf content but does not generate files. The command is as follows:
dcs VarArc view -i ARC \
-t THREADNUM \
-p POSRANGE \
-s SAMPLELIST
The command requires the following parameters to be entered:
ARC: Enter the compressed file path
THREADNUM: Number of threads set
POSRANGE: Set the chromosome name and interval of the query [chr1: 1000-2000]]
SAMPLELIST: Set the sample command list file for query [each line of the file is a sample]]
3. Detailed Tool Parameter Descriptions
3.1 aligner
Supported command-line parameters for aligner:
- --threads: Number of threads. Recommended not to exceed the number of CPU cores.
- --fq1: Input FASTQ1 file. For multiple files, supports comma separation or file lists.
- --fq2: Input FASTQ2 file. For multiple files, supports comma separation or file lists.
- --seqarc-ref: Index file required for decompressing SeqArc format files.
- --read-group: Format is @RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM, where GROUP_NAME is the read group identifier, SAMPLE_NAME is the sample name, and PLATFORM is the sequencing platform.
- --build-index: Build index files required for alignment tools.
- --aln-index: FASTA path required for sequence alignment. Ensure the accompanying index files are in the same directory as the FASTA file.
- --mark-split-hits-secondary: Mark shorter split hits as secondary.
- --use-soft-clipping-for-sup-align: Use soft clipping for supplementary alignment.
- --no-markdup: Do not perform duplicate marking.
- --bam-out: Output BAM file.
- --high-speed-storage: Temporary file storage path. Placing this path on high-speed storage devices can improve performance.
- --fastq-qc-dir: Output directory for FASTQ quality control reports.
- --no-filter: Do not filter FASTQ data.
- --output-clean-fq: Output clean reads.
- --qualified-quality-threshold: Qualified base quality threshold (default: 12).
- --low-quality-ratio: Low-quality base ratio threshold (default: 0.5).
- --mean-qual: Mean quality threshold.
- --quality-offset: Quality value offset. Typical values are 33 or 64. If set to 0, the value is inferred by the program (default: 33).
- --adapter1: Adapter sequence for read1, in uppercase.
- --adapter2: Adapter sequence for read2, in uppercase.
- --adapter-max-mismatch: Maximum allowed base mismatches during adapter search (default: 2).
- --adapter-match-ratio: Minimum matching length ratio for adapters (default: 0.5).
- --trim-adapter: Trim adapters from read sequences instead of discarding reads.
- --min-read-len: Minimum read length threshold (default: 30).
- --n-base-ratio: N base ratio threshold(default: 0.1).
- --trim-ends: Trim a specified number of bases from both ends of the reads, with the format being comma-separated integers.
3.2 bqsr
Supported command-line options for bqsr:
- --threads: Number of threads. Recommended not to exceed the number of CPU cores.
- --in-bam: Duplicate-marked BAM file.
- --out-bam: Path to store the BAM file after base quality score recalibration. If this parameter is omitted, only the recalibration table is generated.
- --reference: Reference genome FASTA file. Must be the same as the one used in the sequence alignment stage.
- --recal-table: Output file containing information for base quality score recalibration.
- --known-sites: Single nucleotide polymorphism database data and known variant sites file. Supports text and gz compressed formats.
- --interval: BED file.
- --interval-padding: Number of bases to pad on both sides of the BED interval.
3.3 variantCaller
Detailed parameter descriptions for variantCaller:
- -I, --input
<file1><file2>...: Input BAM file paths. Multiple BAM paths can be accepted, separated by spaces. - -O, --output
<file>: Output VCF/GVCF BGZF file. The program automatically creates the corresponding index file. - -R, --reference
<file>: Reference genome FASTA file path. - --recal-table
<file>: Input file containing information for base quality score recalibration. - -H, --help: Print help information.
- --version: Print version number.
- -L, --interval
<file>: Target interval BED file path. - -P, --interval-padding
<int>: Number of bases to extend on both sides of the interval. Must be a non-negative integer (default: 0). - -Q, --base-quality-score-threshold
<int>: Base quality score threshold. Bases with quality scores below this threshold will be set to the minimum value of 6 (default: 18). - -D, --max-reads-depth
<int>: Maximum number of reads retained per alignment start position. Reads exceeding this threshold will be downsampled (default: 50). - -G, --gvcf-gq-bands
<int1><int2>...: Reference confidence model GQ groups. Input multiple integers separated by spaces (default: [1,2,3,...,59,60,70,80,90,99]). - -t, --threads
<int>: Number of threads used by the program, ranging from 1 to 128 (default: 30). - --pcr-indel-model
<str>: PCR indel model. Parameter range: {NONE, HOSTILE, CONSERVATIVE, AGGRESSIVE} (default: CONSERVATIVE). - --emit-ref-confidence
<str>: Run the reference confidence scoring model. Parameter range: {NONE, BP_RESOLUTION, GVCF}. NONE: Do not run the reference confidence scoring model; BP_RESOLUTION: Run the reference confidence scoring model, outputting a variant record for each site; GVCF: Run the reference confidence scoring model, grouping reference confidence sites (ALT=<NON_REF>) by GQ value according to the -G parameter (default: NONE). - --compression-level
<int>: Output VCF/GVCF BGZF file compression level, ranging from 0 to 9 (default: 6).
3.4 genotyper
The genotyper processes single-sample GVCF files, outputting VCF files containing only variant records. For joint calling of multiple GVCF files, use the high-performance DPGT software. Detailed parameter descriptions for genotyper:
- -I, --input
<file>: Input GVCF file path. - -O, --output
<file>: Output VCF BGZF file containing genotypes. The program automatically creates the corresponding index file. - -s, --stand-call-conf
<float>: Variant quality score threshold. Variants with quality scores greater than or equal to this value will be output (default: 10.0). - -D, --dbsnp
<file>: dbSNP file. - -t, --threads
<int>: Number of threads used by the program (default: 6). - --version: Print version number.
- -h, --help: Print help information.
3.5 jointcaller (DPGT)
jointcaller is a wrapper for the DPGT tool, used to conveniently run DPGT in standalone mode. DPGT is a distributed joint calling tool. Its detailed parameter descriptions are as follows:
- -i, --input
<FILE>: Input GVCF file list, a text file with one GVCF path per line. - --indices
<FILE>: Optional parameter. The program defaults to searching for index files by appending .tbi to the GVCF path. This parameter can specify the index file path, requiring a text file with one index path per line, matching the order of GVCF paths. - --use-lix
<Boolean>: Use LIX index format. LIX files are simplified index files implemented by DPGT. TBI indexes can be converted to LIX indexes using the tbi2lix program. LIX indexes are approximately 1/80 the size of TBI indexes. Options: true, false (default: false). - --header
<FILE>: Optional parameter. Use a precomputed VCF header file to avoid repeated calculations. - -o, --output
<DIR>: Output folder. - -r, --reference
<FILE>: Reference genome FASTA file. - -l, --target-regions
<STRING>: Target region string or BED file. Defaults to processing the entire genome. - -j, --jobs
<INT>: Number of parallel tasks (default: 10). - -n, --num-combine-partitions
<INT>: Number of partitions during the variant merging phase. Defaults to the square root of the sample count, rounded down (default: -1). - -w, --window
<INT>: Region size processed by each merge-genotype calculation (default: 300M). - -d, --delete
<Boolean>: Whether to delete temporary files. Options: true, false (default: true). - -s, --stand-call-conf
<FLOAT>: Quality score threshold. Variant records with quality scores greater than or equal to this value will be output (default: 30.0). - --dbsnp
<FILE>: dbSNP VCF file path, used for annotating dbSNP IDs. - --use-old-qual-calculator
<Boolean>: Whether to use the old quality score calculator. When set to true, the old calculator is used, matching the results of GATK v4.1.0 with --use-old-qual-calculator true --use-new-qual-calculator false. When set to false, the new calculator is used, matching the results of GATK v4.1.1 and later versions. Options: true, false (default: true). - --heterozygosity
<FLOAT>: Prior probability of heterozygous SNP variants (default: 0.001). - --indel-heterozygosity
<FLOAT>: Prior probability of heterozygous INDEL variants (default: 1.25E-4). - --heterozygosity-stdev
<FLOAT>: Standard deviation of prior probabilities for SNP and INDEL variants (default: 0.01). - --max-alternate-alleles
<INT>: Maximum number of alternate alleles. For variants exceeding this number, the program retains only the most probable alleles up to this count (default: 6). - --ploidy
<INT>: Ploidy (default: 2). - -h, --help: Print help information and exit.
- -V, --version: Print version number and exit.
3.6 SeqArc
3.6.1 Index Creation Functionality, Subcommand index
Parameters for creating indexes:
- -h, --help: Get help information for index creation parameters.
- -r, --ref
<file>: Set the path to the input reference sequence file (FASTA). - -K, --clsdb
<dir>: Set the directory containing reference sequence files for multiple species. The directory must contain names.dmp and nodes.dmp, which can be downloaded from: http://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz. - -H, --hitlimit
<int>: Set the minimizer hit count for taxonomy (default: 2). - -t, --thread
<int>: Set the number of threads (default: 8). - -o, --output
<dir>: Set the output directory for storing generated index files for the reference sequence. - --aligntype
<int>: Set the alignment method for generating index files (default: 2: 1-hash, 2-minimizer, 3-longseq).
If users know the species of the fastq file, they can use the -r parameter to directly select the corresponding reference sequence to generate the index file, and use the -o parameter to save the generated index file. This index file can then be used for compression and decompression. If users do not know the species of the fastq file, they can use the -k parameter to create a species identification library index for all reference sequences in the directory. Then, for each reference sequence, they can use the -r parameter to create the corresponding index file. In this case, the directory specified by -o must be the same as the input directory of -K.
3.6.2 Compression Function, Subcommand compress
The compression parameters are as follows:
- -h, --help: Get help instructions for compression parameters.
- -r, --ref
<file>: Set the path of the input reference sequence. The corresponding index file must exist in this path. - -K, --clsdb
<dir>: Set the directory containing reference sequence files for multiple species. This directory must contain the generated species identification library file and the index files for each species. - -H, --hitlimit
<int>: Set the minimizer hit count for taxonomy, default is 2. Keep this consistent with the creation parameters. - -J, --clsmin
<float>: Set the minimum hit rate for species identification, default is 0.6. - -L, --clsnum
<int>: Set the number of reads to be read from the fastq file for species identification, default is 10000. - -t, --thread
<int>: Set the number of threads, default is 8. - -f, --force: Force overwrite existing files with the same name.
- -i, --input
<file>: Set the input fastq file. - --pipein: Specify if the input is pipeline data.
- -o, --output
<file>: Set the output file path. The .arc suffix will be automatically added. - --blocksz
<int>: Set the size of each data block during compression, default is 50. - -ccheck: After compression is completed and data is written to disk, perform a decompression check to ensure data integrity.
- --calcmd5: Calculate the MD5 of the input file during compression and save it to the compressed file.
- --aligntype
<int>: Set the compression alignment method, default is 2. - -a, --archive
<dir>: Set the directory to be compressed and archived. - --slevel
<int>: Set the compression level for sequences, default is 10. - --verify
<int>: Set the verification method for each data block, default is 10 (no verification), 1 (verify the entire block), or 2 (verify name, seq, and qual separately).
3.6.3 Decompression Function, Subcommand decompress
The decompression parameters are as follows:
- -h, --help: Get help instructions for decompression parameters.
- -r, --ref
<dir/file>: Set the path of the input reference sequence. The corresponding index file must exist in this path. Alternatively, specify the species identification library directory, which must contain the index files for each species. - -t, --thread
<int>: Set the number of threads, default is 8. - -f, --force: Force overwrite existing files with the same name.
- -i, --input
<file>: Set the path of the input compressed file. - -o, --output
<file>: Set the path of the output decompressed file. - -rawtype: Ensure the decompressed output file format matches the original compressed file format. This only applies to mgz format files.
- -dcheck: After decompression is completed and data is written to disk, read the decompressed data blocks and verify them to ensure integrity.
- -x, --extract: Extract one or more files from the compressed archive. The input file must be generated by -a or --archive.
- -l, --list: Display the file information in the compressed archive. The input file must be generated by -a or --archive.
- --show: Display the text MD5 value of the compressed file. This requires --calcmd5 to be specified during compression.
3.7 VarArc
3.7.1 Compression function, subcommand [compress]]
-h, -- help: Get help instructions for using compression parameters
-I, -- input<file>: Set the input vcf file
-o, -- output <file>: Set the output compressed file path
-t, -- thread <int>: set the number of threads, the default is [4]]
-- rowcnt: Set the number of rows for each large block.
-- blkcolcnt: Set the number of samples per block
3.7.2 Decompression function, subcommand [decompress]]
-h, -- help: Get help instructions for using compression parameters
-I, -- input<file>: Set the input compressed file
-o, -- output <file>: set the path of the output decompression file
-t, -- thread <int>: set the number of threads, the default is [4]]
-p, -- pos: Set the chromosome name and range for screening [chr1: 1000-2000]]
-s, -- samplefile: Set the filtered sample name list file, each sample name occupies one line
-S, -- samplelist: Set a list of sample names for filtering, separated by commas.
3.7.3 view function, subcommand [view]]
-h, -- help: Get help instructions for using compression parameters
-I, -- input<file>: Set the input compressed file
-t, -- thread <int>: set the number of threads, the default is [4]]
-p, -- pos: Set the chromosome name and range for screening [chr1: 1000-2000]]
-s, -- samplefile: Set the filtered sample name list file, each sample name occupies one line
-S, -- samplelist: Set a list of sample names for filtering, separated by commas.
3.8 license-proxy
Executable file parameters:
- -h, --help: Print help information.
- --license
<LICENSE_PATH>: Path to the license file. - --bind
<BIND_ADDR>: Listening address. - -d, --daemon: Start in the background.
3.9 report
Organize, visualize, and generate an HTML report for WGS/WES analysis results. Parameters:
- --sample: Sample name.
- --filter: Path to the filter directory.
- --bam_stat: Path to the BAM statistics file.
- --vcf_stat: Path to the VCF statistics file.
- --output: Directory for the output report.
- --is_se: (Optional) Add this flag if single-end sequencing is used.
3.10 lickit
Used to check network connectivity, query the number of available threads for the user, and upload issue logs.
- --ping
<ip:port>: Check network connectivity. - --query
<ip:port>: Query the number of available threads for the user. - --uplog
<ip:port logpath [msg]>: Actively upload issue logs.
3.11 Utility tools
3.11.1 extract-vcf
A standard VCF file contains 8 columns. Columns 6, 7, and 8 occupy significant space but are not required by the program. To reduce IO pressure, this tool extracts only the first 5 columns from the VCF file and replaces the last 3 columns with *. Main parameters:
- --known-sites
<file>: Input VCF file, supporting both text and gz compressed formats. - --output-mini-vcf
<file>: Output VCF file. If the output filename includes gz, it will be in compressed format.
3.11.2 bam-stat
Generate alignment statistics based on the BAM file. In the WGS pipeline, this function is integrated into variantCaller but can also be run separately. Parameters:
- --bam
<file>: Path to the input BAM file. - --bed
<file>: Target regions. For WGS data, specify non-N regions (if unavailable, use the BAM statistics results from variantCaller). For WES data, specify the target region file. - --output
<file>: Path to the output file.
3.11.3 vcf-stat
Generate variant statistics based on the genotyped VCF file, including SNP counts. Parameters:
- --vcf
<file>: Path to the input VCF file, supporting both text and gz formats. Results are output to standard output by default and can be redirected to a specified file (refer to Step 5 in Section 4.1).
3.11.4 tbi2lix
- -i, --input FILE: Input TBI index file.
- -o, --output FILE: Output LIX index file.
- -m, --min-shift INT: Set the minimum interval size for the LIX linear index to 2^INT (default is 14, corresponding to a 16K interval size).
- -h, --help: Print help information and exit.
3.12 Mutect2
This module is a parallel accelerated version of GATK. Use -- nthread to set the number of parallel threads, and use -- java-options "<Xmx and other parameters>" to specify java/jvm parameters. For other parameter descriptions, please refer to GATK
3.13 fastq2vcf
The automation script ($DCS_HOME/libexec/fastq2vcf.sh) In order to facilitate the WGS/WES process construction, the parameters are uniformly transferred to the script to complete the whole process from fastq to vcf. Detailed parameters are as follows
Usage: fastq2vcfsh DCS_HOME=/path/to/dcs-tools refFasta=/path/to/reffa FQ1=/path/to/read1fqgz FQ2=/path/to/read2fqgz [other parameters]
Required parameters:
DCS_HOME: path to the dcs-tools directory
refFasta: path to the reference genome fasta file
FQ1: path to the first FASTQ file
Optional parameters:
FQ2: path to the second FASTQ file (for paired-end data)
proxyHost: proxy host for license authentication (default:127001:8909)
OUTPUT_DIR: output directory (default: current directory)
threads: number of threads to use (default:32)
sample: sample name (default:test)
bed: path to target regions in bed format (optional)
useLowMemIndex: whether to use low memory index (true/false, default:false)
ploidy: sample ploidy (default:2), if greater than 2, java and spark environment is required for genotype calling
dbsnp: path to dbsnp vcf file (optional)
knownSitesFileList: comma-separated list of known sites vcf files for BQSR (optional)
Example:
fastq2vcf
4. Quick Start—Using Example Scripts and Data
After deploying the software as described in Section 1.4, download the quick start data package (cloud platform download link), unzip it, and modify the DCS_HOME and PROXY_HOST values in the example script. DCS_HOME is the path where the software package is unzipped, and PROXY_HOST is the IP and port (default 8909) specified during deployment on the networked node. Execute the corresponding example script for different application scenarios.
tar xzvf dcs-quick-start-xxxx.tar.gz
cd dcs-quick-start
4.1 fastq2vcf
Starting from v2.0, the DCS Tools toolset has added the automatic script of fastq2vcf, which supports the automatic script process of WGS and WES data from fastq to vcf. If you want to customize the process, you can refer to 4.2 and 4.3. Use the method to human regular WGS data example.
$DCS_HOME/libexec/fastq2vcf.sh\
DCS_HOME=<dcs home> \
Ref
FQ1=<r1.fq/gz> \
FQ2=<r2.fq/gz> \
OUTPUT_DIR=<output dir> \
sample=<sample name> \
dbsnp=<dbsnp vcf> \
knownSitesFileList=<some known sites with vcf format seperated by comma>
4.2 WGS
Reference script: quick_start_wgs.sh
#!/bin/bash
# This script provides a quick start guide for using the dcs tool to analyze whole-genome sequencing data.
# The script demonstrates how to build the index of the reference genome, align the reads,
# recalibrate the base quality scores, call variants, and filter the variants.
# if you have any questions, please contact us at cloud@stomics.tech
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
if [ ! -d "$DCS_HOME" ]; then
echo "--- Error: DCS_HOME directory does not exist: $DCS_HOME"
exit 1
fi
if [ ! -r "$DCS_HOME" ]; then
echo "--- Error: No read permission for DCS_HOME: $DCS_HOME"
exit 1
fi
# Before using the dcs tool, you must obtain a license file and start the authentication proxy program
# on a node that can connect to the internet.
# Get the local IP address, which is used for license application.
# The output data field of the following command indicates the IP address.
# curl -X POST https://cloud.stomics.tech/api/licensor/licenses/anon/echo
# start the proxy program
# LICENSE=<path/to/license/file>
# $DCS_HOME/bin/dcs licproxy --license $LICENSE
# Set the following environment variables
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
# Set the following variables for the dcs tool
FQ1=$SCRIPT_DIR/data/fastq/simu.r1.fq.gz
FQ2=$SCRIPT_DIR/data/fastq/simu.r2.fq.gz
FA=$SCRIPT_DIR/data/ref/chr20.fa
KN1=$SCRIPT_DIR/data/known_sites/1000G_phase1.snps.high_confidence.hg38.chr20.vcf.gz
KN2=$SCRIPT_DIR/data/known_sites/dbsnp_151.hg38.chr20.vcf.gz
KN3=$SCRIPT_DIR/data/known_sites/hapmap_3.3.hg38.chr20.vcf.gz
KN4=$SCRIPT_DIR/data/known_sites/Mills_and_1000G_gold_standard.indels.hg38.chr20.vcf.gz
nthreads=8
sample=simu
group="@RG\\tID:sample\\tLB:sample\\tSM:sample\\tPL:COMPLETE\\tCN:DCS"
# run the wgs pipeline ------------------------------------------------------------------------------------
# check the existence of the index files
for file in $FA.0123 $FA.amb $FA.ann $FA.bwt $FA.index1 $FA.index2 $FA.pac $FA.sa;
do
if [ ! -f $file ]; then
echo "File $file does not exist, try to build the index ..."
$DCS_HOME/bin/dcs aligner --threads 16 --build-index $FA
break;
fi
done
# Step 0: Set the working directory
workDir=$SCRIPT_DIR/result_wgs
mkdir -p $workDir
mkdir -p $workDir/${sample}.qcdir
# Step 1: Alignment
$DCS_HOME/bin/dcs aligner --threads $nthreads --aln-index $FA --fq1 $FQ1 --fq2 $FQ2 --read-group $group --no-filter --fastq-qc-dir $workDir/${sample}.qcdir --bam-out $workDir/${sample}.align.bam
# Step 2: base quality score recalibration
$DCS_HOME/bin/dcs bqsr --threads $nthreads --in-bam $workDir/${sample}.align.bam --reference $FA --known-sites $KN1 --known-sites $KN2 --known-sites $KN3 --known-sites $KN4 --recal-table $workDir/${sample}.recal.table
# Step 3: variant calling
$DCS_HOME/bin/dcs variantCaller --threads $nthreads --input $workDir/${sample}.align.bam --output $workDir/${sample}.variantCaller.g.vcf.gz --reference $FA --recal-table $workDir/${sample}.recal.table
# Step 4: genotype calling
$DCS_HOME/bin/dcs genotyper --threads $nthreads --input $workDir/${sample}.variantCaller.g.vcf.gz --output $workDir/${sample}.genotyper.vcf.gz --dbsnp $KN2 -s 30.0
# Step 5: generate the final report
$DCS_HOME/bin/dcs vcf-stat --vcf $workDir/${sample}.genotyper.vcf.gz >$workDir/vcfStat.txt
$DCS_HOME/bin/dcs report --sample ${sample} --filter $workDir/${sample}.qcdir --bam_stat $workDir/bamStat.txt --vcf_stat $workDir/vcfStat.txt --output $workDir/report
After modifying the environment variables in the file, simply run it directly.
# Modify the values of DCS_HOME and PROXY_HOST.
vi quick_start_wgs.sh
sh quick_start_wgs.sh
Examples of the use of polyploid data analysis
export PATH=$PATH:<your spark-submit directory>
$DCS_HOME/libexec/fastq2vcf.sh DCS_HOME=<dcs home> proxyHost=<ip:port of license proxy> refFasta=<ref.That > FQ1=<r1.fq/gz> FQ2=<r2.fq/gz> ploidy=<ploid> OUTPUT_DIR=<output dir>
The GVCF to VCF steps of this script use the jointcaller module of DCS Tools, which is consistent with the results of the old version of GATK (before 4.1.5) by default. If you want to get the results consistent with the new version of GATK (after 4.1.5), you need to use fastq2vcf.The jointcaller script part of sh (as shown below, the last line) is added with the following parameter: -- use-old-qual-calculator false
If ["$useDPGT" = true ]; then
GVCF_FOFN=$workDir/gvcf.fofn
Ls $workDir/${sample}.variantCaller.g.vcf.gz > ${GVCF_FOFN}
$DCS_HOME/bin/dcs jointcaller \
--input ${GVCF_FOFN} \
--output ${workDir} \
--reference ${FA} \
--jobs ${nthreads} \
--memory 1G \
--use-old-qual-calculator false
fi
4.3 WES
The steps are largely the same as WGS, with the only difference being the variantCaller step, which requires an additional interval parameter (-L) specifying the WES bed file.
# Step 3: variant calling
$DCS_HOME/bin/dcs variantCaller --threads $nthreads --input $workDir/${sample}.align.bam --output $workDir/${sample}.variantCaller.g.vcf.gz --reference $FA --recal-table $workDir/${sample}.recal.table -L <wes.target.bed>
4.4 Joint Calling
The reference script is quick_start_jointcalling.sh, similar to WGS. Modify the DCS_HOME and PROXY_HOST environment variables. Additionally, dcs jointcaller requires JAVA_HOME to be set to JDK8 and the spark/bin directory (Spark version 2.4.5 or higher) to be added to the PATH environment variable.
# This script provides a quick start guide for using the dcs jointcaller to perform joint calling on multiple gvcf files
# if you have any questions, please contact us at cloud@stomics.tech
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# dcs jointcaller is a wrapper of DPGT, which is a spark java application
# set JAVA_HOME and add spark/bin to PATH
export JAVA_HOME=/path/to/jdk8
export PATH=/path/to/spark/bin:$PATH
# Before using the dcs tool, you must obtain a license file and start the authentication proxy program
# on a node that can connect to the internet.
# Get the local IP address, which is used for license application.
# The output data field of the following command indicates the IP address.
# curl -X POST https://uat-cloud.stomics.tech/api/licensor/licenses/anon/echo?cliIp=true
# start the proxy program
# LICENSE=<path/to/license/file>
# DCS_HOST=0.0.0.0:8909
# $DCS_HOME/bin/licproxy --command start --license $LICENSE --bind $DCS_HOST --log-level INFO
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
# Set the following variables for dcs jointcaller
# gvcf directory
GVCF_DIR=$SCRIPT_DIR/data/gvcf
# target region
REGION=chr20:1000000-2000000
# reference fasta file
FA=$SCRIPT_DIR/data/ref/chr20.fa
# number of threads
nthreads=4
# java heap max memory
memory_size=8g
# Step 0: Set the working directory
workDir=$SCRIPT_DIR/result_jointcalling
mkdir -p $workDir
# Step 1: Set input file and output dir
# dcs jointcaller input file
GVCF_FOFN=$workDir/gvcf.fofn
ls ${GVCF_DIR}/*gz > ${GVCF_FOFN}
# dcs jointcaller output directory
OUT_DIR=$workDir/result
# Step 2: Run dcs jointcaller
$DCS_HOME/bin/dcs jointcaller \
--input ${GVCF_FOFN} \
--output ${OUT_DIR} \
--reference ${FA} \
--jobs ${nthreads} \
--memory ${memory_size} \
-l ${REGION}
4.5 SeqArc
The reference script is quick_start_seqarc.sh, similar to WGS. Modify the DCS_HOME and PROXY_HOST environment variables.
#!/bin/bash
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
nthreads=2
FQ1=$SCRIPT_DIR/data/fastq/simu.r1.fq.gz
FA=$SCRIPT_DIR/data/ref/chr20.fa
ARCFA=$SCRIPT_DIR/data/arcref/
mkdir -p $ARCFA
workDir=$SCRIPT_DIR/result_seqarc
mkdir -p $workDir
# Step 1: create index
$DCS_HOME/bin/dcs SeqArc index -r $FA -o $ARCFA
# Step 2: compress
$DCS_HOME/bin/dcs SeqArc compress -t $nthreads -r $ARCFA/chr20.fa -i $FQ1 -o $workDir/test
# step 3 : decompress
$DCS_HOME/bin/dcs SeqArc decompress -t $nthreads -r $ARCFA/chr20.fa -i $workDir/test.arc -o $workDir/simu.r1.fq
VarArc
#!/bin/bash
# set the location of the dcs tool
export DCS_HOME=/path/to/dcs
# Set the following environment variables to your desired values
export PROXY_HOST=127.0.0.1:8909
# check the status of the proxy program
$DCS_HOME/bin/dcs lickit --ping $PROXY_HOST
# query the available resources provided by the certification
$DCS_HOME/bin/dcs lickit --query $PROXY_HOST
# Get the directory where the script is located
SCRIPT_PATH=$(realpath "$0" 2>/dev/null || readlink -f "$0" 2>/dev/null || echo "$0")
SCRIPT_DIR=$(dirname "$SCRIPT_PATH")
nthreads=4
VCF=$SCRIPT_DIR/data/vcf/demo.Vcf.gz
WorkDir=$SCRIPT_DIR/result_vararc
Mkdir -p $workDir
# Step 1: compress
$DCS_HOME/bin/dcs VarArc compress -t $nthreads -i $VCF -o $workDir/demo It's okay.arc --rowcnt 100 --blkcolcnt 2000
# step 2 : decompress
$DCS_HOME/bin/dcs VarArc decompress -t $nthreads -i $workDir/demo.Arc -o $workDir/demo.vcf.gz
5. Release Notes
v2.0
Support the variation detection of polyploid species, somatic variation detection part began to support multi-thread acceleration, aligner support new index structure, enhance stability.
| Modular | Type | Depaint |
|---|---|---|
| WGS/WES | Functional Development | aligner: Supports low memory index mode (running speed is slightly reduced), is more friendly to resource requirements, and can solve genome bugs like Cycas variantCaller: Supports the detection of variation in polyploid species (parameter: -- ploidy). Add corresponding statistical fields for WES data report: reports that output WES data. util: the bamStat function (separate statistics for bam) is consistent with that of variantCaller. The Statistics field of WES is added. |
| Somatic cell variation detection | Functional Development | Support multi-thread acceleration mode, with -- nthread parameter setting, 32 threads under the speed can reach about 8 times of the original version. |
| Population Genome conjoint analysis | Functional Development | Support for polyploid species (parameter: -- ploidy) |
| VarArc | Functional Development | Added this module to support compression and decompression of GVCF/VCF files (text or gz format) |
Remarks:
1. bam statistics of WES data do not use the statistical results of variantCaller, but use the bamStat function of util for separate statistics.
2. After the variantCaller, it is recommended to use jointcaller to perform the operation from GVCF to VCF.
v1.5.0
Support using a dongle for offline authentication to enable standalone use of this tool.
| Module | Type | Description |
|---|---|---|
| All Modules | Feature Development | Added support for using a dongle for offline license authentication, allowing DCS Tools to be used on machines without an internet connection. |
| WGS/WES | Bug Fix | genotyper: Fixed the issue of high memory usage. |
v1.4.0
| Module | Type | Description |
|---|---|---|
| WGS/WES | Feature Development | Added the --include-non-variant-sites parameter to the genotyper, which functions similarly to the parameter of the same name in GATK's GenotypeGVCFs. It retains non-variant block information from the GVCF in the VCF and expands it, facilitating downstream genetic disease analysis and similar scenarios. Note that when this parameter is enabled, the reference file (-R) must be specified. |
| SeqArc | Bug Fix | Fixed a bug causing abnormal retrieval of available disk space for users. |
v1.3.0
| Module | Type | Description |
|---|---|---|
| WGS/WES | Bug Fix | aligner: 1. Fixed the issue where overlong fasta lines cause errors in fai file generation. 2. Fixed the destructor bug on ARM Euler OS variantCaller: 1. Fixed segmentation faults in certain scenarios. 2. Fixed memory issues for specific species |
| SeqArc | Feature Dev | Implemented a Windows decompression tool |
| SeqArc | Feature Dev | Supported direct decompression of arc files to gz format, with pipeline mode support |
| SeqArc | Feature Dev | Supported compression and decompression of U-bases |
| Somatic Mutation Detection | Feature Dev | Added this new module |
v1.2.0
Added support for ARM instruction architecture and introduced sample size and data volume (compressed) statistics to align with the license model based on sample size and data volume.
| Module | Type | Description |
|---|---|---|
| WGS/WES | Bug Fix | bbqsr: Fixed a bug when chromosome length exceeds 2G.variantCaller: Fixed a bug with the -L parameter in certain scenarios and resolved gVCF index file issues. |
| Feature Development | aligner: Added shared memory functionality (enable with --useSharedMemIndex parameter).variantCaller: 1. Added 15x coverage statistics. 2. Made compatible with overlapping BED files.report: Updated report version and included 15x coverage statistics. | |
| SeqArc | Feature Development | Implemented a standalone decompression tool. |
| Feature Development | Added gene library statistics functionality. | |
| JointCalling | Bug Fix | Fixed a partition end bug. |
v1.1.0
| Module | Type | Description |
|---|---|---|
| WGS/WES | Bug Fix | Resolved memory leak issue in aligner |
| Bug Fix | Fixed aligner hanging when building indexes for certain ref.fasta files | |
| SeqArc | Feature Development | Implemented cross-platform instruction sets using simde |
| Feature Development | Added standalone decompression tool | |
| Feature Development | Implemented statistical functionality for gene libraries |
v1.0.1
改动模块:aligner, report
| Module | Type | Description |
|---|---|---|
| WGS/WES | Bug Fix | Fixed a coredump issue in the aligner under certain scenarios |
| Bug Fix | Fixed an issue where the aligner output file was a pure filename without a path | |
| Bug Fix | Renamed the bam index file generated by the aligner to *.bam.bai instead of *.bai | |
| Bug Fix | Fixed a potential issue in the cumulative depth frequency plot in the report module | |
| Bug Fix | Added set -e to the WGS script in the quick_start package to exit immediately on error |
v1.0.0
| Module | Type | Description |
|---|---|---|
| WGS/WES | Feature | Added alignment and variant-related statistics functionality. |
| SeqArc | Feature | Restructured the project framework according to the AVS-G standard. |
| Feature | Added compression functionality for long-read data. | |
| Feature | Adjusted parameter parsing, added subcommands, and long/short parameters. |
6. Notes and Common Issues
6.1 WGS/WES
- During alignment, ensure the reference sequence index file is prepared in advance to avoid errors.
- The BQSR step outputs only the recal.table by default. To generate a base quality-recalibrated BAM file, set the --out-bam parameter.
- For WES alignment statistics, there are two methods: –Specify the interval parameter in variantCaller as the WES bed file. –Run the bam-stat tool separately and set the --bed parameter. If no bed file is provided, statistics will default to the whole genome, leading to inaccurate coverage and depth results.
6.2 Joint Calling
- Input GVCF files must include TBI index files.
- The reference genome FASTA file requires FAI and DICT files.
- For large sample sizes, allocate sufficient memory to avoid overflow (refer to the runtime examples in Section 7).
6.3 SeqArc
- For compression, input FASTQ filenames must have the following suffixes: .fq, .fastq, .fq.gz, or .fastq.gz. Otherwise, the file type will be considered invalid.
- Input reference sequence files must be text files with the suffixes .fa or .fasta. Otherwise, the file type will be considered invalid.
- For decompression, input ARC files must be generated by this program. Otherwise, the file type will be considered invalid.
- The reference sequence file used for decompression must match the one used for compression. If they differ, an error will occur: [ref load md5 error].
- If the FASTQ file is incomplete, an error will occur: [read fastq error].
- If the seq and qual lengths are unequal, an error will occur: [seqlen is not equal quallen].
- For compression or decompression, file read/write errors will trigger: [read file err] or [write file err].
- During decompression, if a data block fails verification, an error will occur: [block check fail]. This may indicate compression anomalies or incorrect data storage.
- Set ulimit -n to avoid runtime crashes, which may generate core dumps.
6.4 License Proxy
Tool Issue: DCS Tools or lickit cannot connect to licproxy
If you encounter error messages like:
connection refusedcouldn't connect to server
Please follow these troubleshooting steps:
- On the machine running DCS Tools or lickit, check connectivity to the License proxy using:
curl -v http://$PROXY_HOST/api/licensor/licenses/anon/pool
Replace $PROXY_HOST with your licproxy's internal IP address and port (e.g., 172.20.9.149:8909), NOT the public IP address you used when obtaining the License file!
If DCS Tools/lickit and licproxy are on the same machine, you may use 127.0.0.1:8909 for $PROXY_HOST
The expected return should include Success. If it prompts that the connection cannot be established, please proceed to check whether the License proxy process exists by following the steps below.
- Check whether the process exists on the machine where licproxy is deployed
ps aux | grep licproxy
If the process does not exist, please follow the steps in Section 1.4 Software Deployment to proceed. If the process exists, contact your IT operations team to ensure that the machines running DCS Tools or lickit can access the machine where licproxy is located as described in Step 1. If the network connection is unavailable, request your IT operations team to enable it.
- Check whether the script or command line for submitting tasks correctly sets the variable PROXY_HOST
Please verify whether the script or command line you are using overrides the environment variable PROXY_HOST. For example, if licproxy is deployed at 172.20.9.149:8909, ensure you set the following before running the script or command line:
export PROXY_HOST=172.20.9.149:8909
./path/to/your
# or
PROXY_HOST=172.20.9.149:8909 /path/to/your
Common License Authentication Error Messages :
- Permission denied (No permission or authentication failed)
- Size must be greater than 0 (The number of submitted threads must be greater than zero)
- Pool size exceeded (The number of submitted threads exceeds the current thread limit)
- License expired (The license has expired)
- Unknown error (Unknown error)
7. Performance and Runtime
7.1 WGS
On an Alibaba Cloud ECS i4g.8xlarge instance (32 cores, 128GB RAM) with SSD storage, the DCS tools completed the FASTQ-to-VCF process in the following times for different datasets:
| Data | Runtime(h) | Threads | Max Memory (GB) |
|---|---|---|---|
| HG001 30X NovaSeq | 1.82 | 32 | 101 |
| HG001 30X DNBSEQ-G400 | 1.91 | 32 | 100 |
| HG005 40X DNBSEQ-G400 | 2.45 | 32 | 120 |
For the low-memory index aligner version, test the HG001 30X WGS NovaSeq data on the Intel(R) Xeon(R) Gold 6442Y model.
| Tools | Running time (h) | Number of threads | Maximum memory (GB) |
|---|---|---|---|
| aligner | 0.83 | 32 | 97.99 |
| aligner(low memory index) | 1.11 | 32 | 39.22 |
7.2 SeqArc
On an Alibaba Cloud ECS c6a.xlarge instance (4 cores, 8GB RAM), compression and decompression tests were performed on NA12878 (30X WGS) data.
Compression Results:
| Input Data | Runtime | Compression Ratio (vs. gz) |
|---|---|---|
| r1.fq.gz(27G) | 27m 24s | 5.17 |
| r2.fq.gz (30G) | 29m 14s | 4.10 |
Decompression Results:
| Input Data | Runtime |
|---|---|
| r1.fq.arc | 14m 32s |
| r2.fq.arc | 14m 25s |
7.3 Joint Calling
Performance tests on the 1KGP dataset and internal large-scale population datasets showed the following results for dcsjointcaller (DPGT):
| Data | Samples | Processes (6GB RAM each) | Core Hours | Runtime |
|---|---|---|---|---|
| 1KGP | 2504 | 256 | 6963 | 27.2 hours |
| Internal 10K | 9165 | 256 | 21376 | 83.5 hours |
| Internal 52K | 52000 | 4400 | 407760 | ~6 days |
| Internal 105K | 105000 | 3675 | 1026155 | ~19 days |
7.4 VarArc
In the local linux cluster, the performance of VarArc is tested on the population variation VCF data (from The CKB project) as shown in the following table:
| Name | Data source | gz Data size/GB | Metacentric | Number of samples | Bit number | Field Characteristics | Tools | comp_size(GB) | Compression ratio | comp_RealTime(s) | comp_CPUTime(s) | comp_CPUUsage | comp_MaxRAM(GB) | Compression speed (With gz as numerator) MB/s | decomp_RealTime(s) | decomp_CPUTime(s) | Decomp_CPUUsing | decomp_MaxRAM(GB) | Decompression speed (With gz as numerator) MB/s |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| data1 | CKB | 15.00 | chr1 | 6w | 8w | Variation contains fields such as GT,AD,DP, etc. | VarArc | 6.32 | 2.37 | 1251 | 31221 | 2510% | 29.2 | 12.3 | 352 | 5091 | 1466% | 7.4 | 43.7 |
| bcftools | 18.80 | 0.80 | 510 | 4630 | 916% | 0.2 | 30.1 | 357 | 3745 | 1058% | 0.1 | 43.0 | |||||||
| data2 | CKB | 25.90 | chr10 | 10.5w | 8w | Variation contains fields such as GT,AD,DP, etc. | VarArc | 11.17 | 2.32 | 1783 | 48871 | 2754% | 94.39 | 14.9 | 587.31 | 8533.3 | 1468% | 9.5 | 45.2 |
| bcftools | 32.33 | 0.80 | 832 | 7933 | 960% | 0.16 | 31.9 | 606.16 | 6713.36 | 1116% | 0.1 | 43.8 | |||||||
| data3 | CKB | 60.70 | chr22 | 10.5w | 524w | Variation with GT field only | VarArc | 53.79 | 1.13 | 13423 | 372176 | 2778% | 31.03 | 4.6 | 3881.66 | 108601.14 | 2815% | 3.2 | 16.0 |
| bcftools | 52.40 | 1.16 | 10189 | 39974 | 397% | 0.06 | 6.1 | 8186.85 | 43343.32 | 535% | 0.1 | 7.6 |
In the local linux cluster, the performance of VarArc is shown in the following table on a single sample of gVCF data (from The CKB project):
| gz Data size/GB | Tools | comp_size(GB) | Compression ratio | comp_RealTime(s) | comp_CPUTime(s) | comp_CPUUsage | comp_MaxRAM(GB) | Compression speed (With gz as numerator) MB/s | decomp_RealTime(s) | decomp_CPUTime(s) | Decomp_CPUUsing | decomp_MaxRAM(GB) | Decompression speed (With gz as numerator) MB/s |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 51.6 | VarArc | 15.52 | 3.3 | 4141.1 | 112007.3 | 2690% | 14.1 | 12.8 | 1213.4 | 10153.0 | 831% | 8.9 | 43.6 |
| bcftools | 54.25 | 1.0 | 2438.3 | 10293.2 | 436% | 0.0 | 21.7 | 1474.8 | 7947.5 | 570% | 0.0 | 35.8 |
7.5 Mutect2
WES data (about 11G bases for normal and tumor)
| Edition | Running time |
|---|---|
| gatk-Mutect2 | 5824 s |
| dcs-Mutect2 (32 threads) | 726 s |
WGS数据(normal:221G bases,tumor:642G bases)
| Edition | Running time |
|---|---|
| gatk-Mutect2 | 57.3 h |
| dcs-Mutect2 (32 threads) | 7.5 h |
8. FAQ
8.1 Deployment-Related Questions
1. What are the platform requirements for DCS Tools? DCS Tools is designed as an executable program running on Linux systems.
The recommended Linux distributions are: CentOS 7, Debian 8, Ubuntu 14.04, and later versions.
2. What are the hardware requirements for the system? The CPU should have at least 8 cores, and the memory should be at least 128GB. It is also recommended to use SSDs with faster read/write speeds.
3. Is an internet connection required during the software deployment process? DCS Tools performs license validation during runtime to ensure legality. Therefore, one machine with internet access (to communicate with the validation server) is required. Other machines (typically compute nodes) do not need internet access but must be able to communicate with the internet-connected machine. Of course, a single internet-connected machine can also deploy the software.
4. What files are needed for software deployment? The software package + license file can be obtained through the cloud platform (cloud@stomics.tech) or a sales manager.
We offer a 30-day free trial. Please contact a sales manager for details.
5. After successfully deploying the license proxy, why do license-related errors still occur when running DCS Tools? After deploying the license proxy, you can use the commands provided in the documentation to confirm whether the connection to the cloud platform is successful. Additionally, each license has a corresponding upper limit on the number of threads. If this limit is reached, new tasks will fail to start. You will need to wait for running tasks to complete or expand the thread limit.
8.2 Tool-Related Questions
1. Does joint calling output a merged gvcf or a vcf? Since the traditional method of generating a multi-sample gvcf file in one go requires significant computational resources and storage space, we have not adopted this strategy to obtain gvcf results for all samples. We recommend that users retain the gvcf files for individual samples. If joint calling is required for specific samples later, it can be run directly based on the single-sample gvcf files.
2. Does WGS support the analysis of non-diploid species? The variant detection workflow of DCS Tools is built based on a diploid detection model. Therefore, WGS currently cannot perform effective variant detection for polyploid samples.
3. Why does the runtime for WGS data significantly exceed the time stated in the documentation? In addition to data volume, the runtime of the WGS workflow is also constrained by the read/write speed of the storage device. In our testing environment, we used high-performance SSDs to minimize the impact of this factor on workflow runtime.
4. What platform's data was used for the compression tests? The compression method used by DCS Tools has already compressed tens of petabytes of data in practice. During the large-scale testing conducted in the development phase, the most representative dataset was the AVS-G reference test set. This dataset was extracted from the NCBI-SRA database and covers 13 of the most representative species, including 39 sets of FASTQ data. It comprehensively covers various scenarios on NCBI in terms of sample attributes, experimental methods, sequencing technologies, sequencing generations, and sequencing platforms.
As a reference, the compression ratio of DCS Tools on this test set was 8.11, while gzip achieved a compression ratio of 4.15. Given that different attributes such as species and sequencing platforms can affect the compression ratio, users are advised to make decisions based on actual test results of DCS Tools and similar tools on their own data.
Detailed information about the AVS-G reference test set can be obtained by contacting a sales manager or reaching out to us through the official account.
5. How does DCS Tools compare to the commonly used pigz when compressing 100GB of FASTQ source files? In a 32-thread environment, tests were conducted on 100GB of human WGS paired-end data. The results are as follows: | Data | pigz Compression Ratio | pigz Compression Speed | pigz CPU Usage | DCS Tools Compression Ratio | DCS Tools Compression Speed | DCS Tools CPU Usage | | ---------------------- | ------- | -------- | ----------- | ------------ | ------------- | ---------------- | | BGI-SEQ500(ERR2755197) | 3.93 | 450MB/s | 3224% | 5.66 | 937MB/s | 3177% | | NovaSeq(SRR10965088) | 4.9 | 220MB/s | 3175% | 21.15 | 794MB/s | 3147% |
6. Will the compression ratio vary for different FASTQ files from the same platform? What are the main reasons? FASTQ files primarily consist of sequencing sequences and quality scores, and the differences arise from these two components.
First, for sequencing sequences, DCS Tools recommends using a mode that includes a reference sequence during compression and decompression, which can effectively improve the compression ratio. The reference sequence is the genome sequence of the species. However, this mode is less effective for transcriptome sequencing and difficult to use for metagenomic sequencing. Even for genomic sequencing, the accuracy of sequencing varies across platforms/models. Generally, the more accurate the sequencing, the better the compression ratio.
Next, for quality scores, the types of quality values differ across models. Quality values are generally divided into two types: 40 and 4. Clearly, fewer types result in a better compression ratio, which is the main source of compression ratio differences. Additionally, quality scores themselves are highly random, which is strongly related to the sequencer. The more random the data, the worse the compression ratio.